一种基于Fe3O4的磁性固体酸催化剂及其制备方法、应用与流程
本发明涉及磁性纳米催化剂技术领域,具体涉及一种基于fe3o4的磁性固体酸催化剂及其制备方法、应用。
背景技术:
皂素是一种药用价值极高的医药中间体,是生产蛋白同化激素、肾上腺皮质激素、性激素、孕激素等四大类的甾体激素药物的不可替代原料,被誉为“药用黄金”。皂素主要由薯蓣科植物(黄姜、穿山龙等)根茎中所含薯蓣皂苷经酸水解获得。薯蓣皂素具有降脂、溶血、消炎等药理作用。但是经研究表明薯蓣科植物原料中皂苷含量仅为1%~2%,并且传统的方法均采用无机强酸水解均相反应法,共存问题是提取量少、强酸腐蚀反应设备,不便于分离、工艺复杂不环保,造成极大的环境污染,最重要是薯蓣科植物里面含有较高含量的糖、淀粉、纤维等物质,在强酸水解皂苷的时候,容易被破坏而无法利用。开发出高效而清洁的皂素生产工艺显得至关重要,而解决皂苷水解污染以及提高皂素提取率是其中突破的关键点。用固体酸代替常用的无机酸是解决水污染的重要途径。尽管国内外在固体酸的理论和应用研究取得了较大的研究进展,但其工业应用研究还有待进一步深入,特别是针对具有特殊用途的生物质的水解研究的较少。
技术实现要素:
本发明的目的在于,针对上述现有技术的不足,提出一种基于fe3o4的磁性固体酸催化剂及其制备方法、应用,该种催化剂解决了传统的有机酸水解存在的不足之处,它可以避免污染环境,腐蚀设备;并且具有很好分散性,不易被氧化和耐碱性,稳定性好,循环利用损失小。
本发明提出一种基于fe3o4的磁性固体酸催化剂,具有以下结构式:
一种基于fe3o4的磁性固体酸催化剂的制备方法,包括以下步骤:
s11:将fe3o4颗粒,超声分散于乙醇和水溶液中,并加入檬酸三钠,剧烈搅拌,回流1-3h,冷却后,用磁铁将fe3o4颗粒从反应液中分离出来,然后进行洗涤、真空干燥,待用;
s12:将三乙烯四胺2-(3,4环氧环己基)乙基三甲氧基硅烷溶(etms)液混合加到干燥的甲苯溶液,回流加热,机械均速搅拌,20h-24h后,加入s11处理后的fe3o4颗粒和乙醇溶液,加完之后继续搅拌20h-24h,反应完毕后浸泡洗涤干燥,得到中间产物;
s13:将s12合成的中间产物,在冰浴条件下用氯磺酸磺化3-5h,得到磁性固体酸fe3o4@etms-teta-so3h。
进一步地,s11中的fe3o4颗粒质量、乙醇体积和水溶液体积比为10-20:1-50:10-50。
进一步地,s11中的fe3o4颗粒与柠檬酸钠的质量比为10-20:1-5。
进一步地,s12中四乙烯三胺的体积、2-(3,4环氧环己基)乙基三甲氧基硅烷溶液的体积和fe3o4颗粒的质量体积比为1-5:9-13:5-6。
进一步地,s12中的得到的中间产物分别依次经过甲醇、乙醇和水溶液进行浸泡洗涤。
进一步地,s13中的中间产物的质量与氯磺酸的体积比为0.1-1:0.1-2。
一种提取皂素的方法,其特征在于:包括以下步骤:
s21:将薯蓣科植物根茎洗净粉碎过筛,取筛下物,加入乙醇,并在回流状态下提取,待布氏漏斗抽滤后,用乙醇洗涤,合并滤液,旋干液体,得到的固体烘干即得皂苷粗品;
s22:将皂苷粗品置于配有冷凝管及搅拌装置的三口烧瓶内,在机械搅拌条件下加入磁性固体酸催化剂、乙醇,在温度为80-110℃下反应4h-6h,冷却至室温,用磁铁回收磁性固体酸,加入乙醇洗涤磁性固体酸且待收集完后,洗涤液和剩余反应液加热旋转蒸干,得到固体产物;
s23:向s22所得固体产物采用索式提取法,用有机溶剂进行抽提,蒸发浓缩得到皂素。
进一步地,s21中的薯蓣科植物的和乙醇的体积比为1:1-10;s22中皂苷粗品的质量、磁性固体酸催化剂的质量和乙醇的体积比为0.1-0.5:0.1-1.5:5-20。
进一步地,s23中的有机溶剂为石油醚、氯仿或二氯甲烷。
本发明制备的磁性固体酸具有酸催化能力和磁控分离的双重特性,符合绿色化学化工的发展理念。与现有技术相比,其合成反应方法简单、反应条件温和、操作安全、对设备无腐蚀,另外与传统酸水解皂苷方法相比,本发明先集中提取皂苷,再加制备的磁性固体酸醇解提取皂素,溶剂可回收利用,无酸性废液排放。因此它具有广泛的工业应用的前景。
本发明的一种基于fe3o4的磁性固体酸催化剂及其制备方法、应用,与现有技术相比,本发明的有益效果为:
1)本涉及的合成新固体酸方法简单、反应条件温和、操作安全,对设备无腐蚀,对环境友好,且水解黄姜皂苷活性高,实用性强,适合推广应用。
2)本发明所述磁性固体酸催化剂水解活性效率比同质量的硫酸水解高。
3)本发明在优化提取皂素清洁工艺,先将从黄姜粗粉中集中提取黄姜皂苷元,再利用制备的固体酸,按照不同比例醇解皂苷元,得到薯蓣皂素。
4)本发明制备了一种新的磁性固体酸催化剂,并应用于薯蓣科植物等中皂素的提取。磁性固体酸提取效率高,可循环利用,并可解决采用无机强酸水解造成的废液酸度高、色度大、盐份高、组成复杂等问题,具有显著的经济和环境效益。
说明书附图
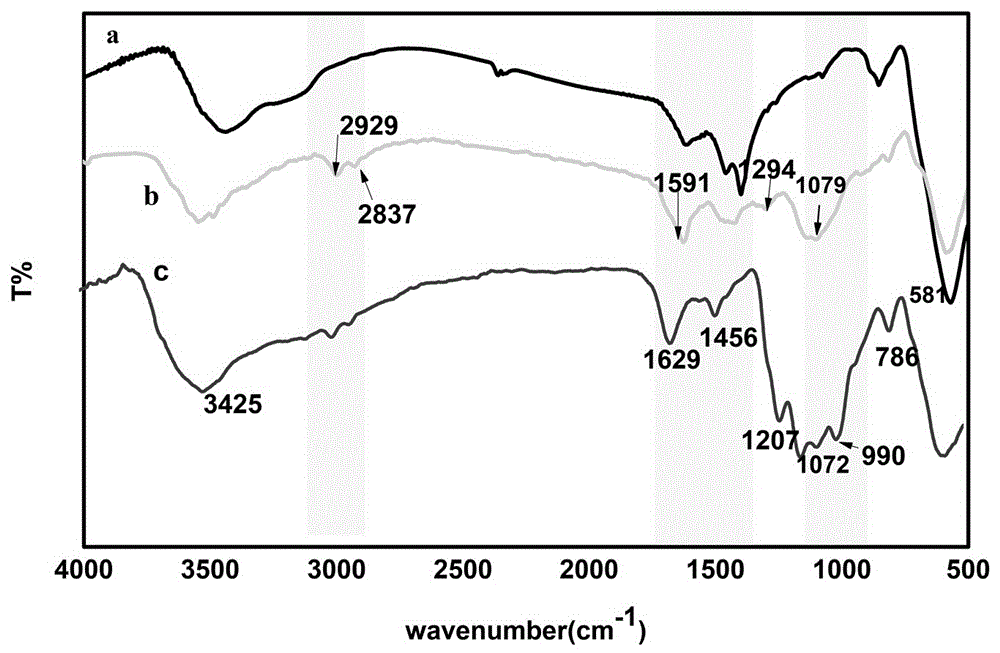
图1为本发明一种基于fe3o4的磁性固体酸催化剂的红外光谱图;
图2为本发明一种基于fe3o4的磁性固体酸催化剂的热重分析图。
具体实施方式
为下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外,本领域技术人员对本发明所做的各种改动或修改,这些等价形式同样落于本申请所要求保护的范围内。本发明实施例中的配比均为以重量计。
实施例1
一种基于fe3o4的磁性固体酸催化剂,其制备方法包括如下步骤:
1)以fe3o4颗粒为原料,加入柠檬酸三钠煮沸;具体包括如下步骤:称取fe3o4颗粒(45.0g)超声10min分散于100ml无水乙醇与20ml蒸馏水混合溶液中,80°c下机械搅拌4h;用磁铁分离出fe3o4颗粒,分别用蒸馏水和乙醇洗涤三次至ph为中性,60℃下真空干燥;
2)量取5ml2-(3,4环氧环己基)乙基三甲氧基硅烷溶液加入100ml干甲苯、10ml四乙烯三胺的搅拌溶液中,混合物在80°c机械搅拌24h。称取步骤1)制备的fe3o4质量为1.1802g,和10ml乙醇加入到反应液中,解决方案是在80°c搅拌24h,待反应完毕fe3o4@sio2-ep-teta在外部磁铁磁作用下分离之后甲醇和乙醇洗。然后,将其与乙醇浸泡24小时,除去未反应的底物和副产物,再分别用蒸馏水和乙醇洗涤三次至中性,60℃下真空干燥得到最终产品fe3o4@etms-teta质量为3.6254g
3)称量步骤2)中的fe3o4@etms2-teta颗粒1.1706g超声分散于40ml氯仿,二氯甲烷的其中的一种或两种混合液中,滴加入氯磺酸(0.5ml),滴完后继续反应6h,反应过程中产生的hcl气体用naoh溶液吸收;反应结束后用磁铁分离出fe3o4@etms-teta@so3h,用无水ch2cl2,无水乙醇洗涤多次,真空干燥,烘干称其重量为1.3031g即得磁性固体酸催化剂。
将本实施例不同步骤所得产物分别进行红外光谱分析,结果分别见图1;第一步骤柠檬酸三钠修饰的fe3o4颗粒572cm-1特征峰为fe-o伸缩震动、3500左右的为纳米颗粒表面羟基-0h的峰。第二步骤fe3o4@etms-teta的红外光谱图。800cm-1附近的谱带表示归属于si-o伸缩振动,根据所制备出四氧化三铁连接硅烷偶联剂红外图谱可以看出1079cm-1处的吸收峰由si-o(h)伸缩振动和si-0-si的不对称伸缩振动引起,2929cm-1和2837cm-1c–h处的峰值归因于脂肪族c-h伸缩振动,是亚甲基峰,1591cm-1和1294是c-c和c-n的拉伸吸收峰,而且环氧基的红外吸收峰在910cm-1处,但是b中没有,说明四乙烯三胺(与2-(3,4环氧环己基)乙基三甲氧基硅烷发生了开环反应。步骤三磺化纳米颗粒上3440cm-1和1632cm-1处表现出两个特征吸附带,与磺酸基(so3h)的so-h键的o=h拉伸和弯曲振动有关。拉伸和拉伸酸性o-h基团的平面外弯曲为两个宽带分别在2925cm-1和960cm-1,对应于si-o-si的带拉伸出现在1072cm-1,而吸收带在1456cm-1(对应于不对称的so2拉伸)和990-1271cm-1范围内的宽峰(归因于对应于si-o-si,fe-o-si的重叠对称so2伸展)表明mnp具有so3h组。,红外光谱清楚地表明所合成的催化剂fe3o4@etms-teta表面被磺酸基团成功功能化。
下面是图2中a、b、c分别为fe3o4、fe3o4@etms-teta和fe3o4@etms-teta@so3h在40-800℃下随温度升高逐渐失重的热重图。从图中可以看出,各个样品均随温度升高而重量逐渐降低,fe3o4在250℃的失重归属于表面羟基脱水逸出,对于fe3o4@etms-teta纳米颗粒,低于250℃时第一次减重15%源于表面残余水损失,fe3o4@etms-teta在300℃到700℃左右的第二次重量损失为催化剂外层有机物的分解,主要是分解除去以化学键合结合的四乙烯三胺和2-(3,4-环氧环己基)乙基三甲氧基硅烷形成有机物质,fe3o4@etms-teta@so3h与fe3o4@etms-teta比较而言在200到500℃时失重严重,这是因为磺酸基团的热分解,第三第四次失重是因为表面残留有机物的分解。从tga曲线中的重量损失百分比来看,fe3o4,fe3o4@etms-teta,fe3o4@etms-teta@so3h纳米颗粒的重量损失分别为7.94%,24.78%,42.28%左右,说明fe3o4表面物质被成功合成包覆,且催化剂被成功引入磺酸基团,在fe3o4表面包覆物质的接枝密度百分比约为16.84%,催化剂都是在150℃下发生热分解,表现出良好的热稳定性。
实施例2
实施例2所述的步骤1)修饰fe3o4颗粒,以及步骤3)磺化反应与实施例1相同,不同之处在于:步骤2)fe3o4@etms-teta颗粒的制备条件不同。其制备方法如下:
量取5ml2-(3,4环氧环己基)乙基三甲氧基硅烷溶液加入100ml干甲苯、10ml四乙烯三胺的搅拌溶液中,混合物在80°c机械搅拌24h。称取步骤1)制备的fe3o4质量为1.8559g,和15ml乙醇加入到反应液中,在80°c下搅拌24h,待反应完毕fe3o4@etms-teta,用外部磁铁磁作用下分离,用甲醇和乙醇洗涤。然后,再在乙醇中浸泡24小时,除去未反应的底物和副产物,再分别用蒸馏水和乙醇洗涤三次至中性,60℃下真空干燥得到最终产品fe3o4@etms-teta质量为8.2141g
3)称量步骤2)中的fe3o4@etms-teta颗粒1.0326g超声分散于60ml氯仿,二氯甲烷的其中的一种或两种混合液中,然后在冰浴下滴加氯磺酸(0.5ml,15分钟滴完),滴完后继续反应6h,反应过程中产生的hcl气体用naoh溶液吸收;反应结束后用磁铁分离出fe3o4@etms-teta@so3h,用无水ch2cl2洗涤四次,再用无水乙醇洗涤两次至中性,在60℃下真空干燥,烘干称其重量为1.2934g即得磁性固体酸催化剂
实施例3
实施例3所述的步骤1)修饰fe3o4颗粒以及步骤2)fe3o4@etms-teta颗粒的制备条件与实施例1相同,不同之处在于:步骤3)中颗粒磺化的制备条件不同。
称量步骤2)中的fe3o4@etms-teta颗粒1.0733g超声分散于60ml无水ch2cl2中,然后在冰浴下滴加氯磺酸(0.6ml),滴完后继续反应7h,反应过程中产生的hcl气体用naoh溶液吸收;反应结束后用磁铁分离出fe3o4@etms-teta@so3h,用无水ch2cl2洗涤四次,再用无水乙醇洗涤两次至中性,在60℃下真空干燥,烘干称其重量为1.3658g即得磁性固体酸催化剂。
实施例4
将实施例1所得磁性固体酸催化剂应用于黄姜中的皂素提取,具体包括如下步骤:
1)将干燥黄姜用破碎机破碎后过200目筛,取筛下物与无水乙醇以1:10(质量与液体体积比)在回流状态下提取6小时,布氏漏斗抽滤后,用乙醇洗涤,合并滤液,旋干液体,得到的固体烘干,即得皂苷;
2)称量干燥的黄姜皂苷0.2030g,量取15m无水乙醇,加入到100ml的三口烧瓶。机械搅拌10min后,加入固体酸fe3o4@etms-teta-so3h0.5436g,回流搅拌5小时,用磁铁分离固体酸,液体倒进单口烧瓶旋转蒸发浓缩至干,所得固体,用120ml氯仿索式抽提5小时,然后旋转蒸发至干,用石油醚溶解定容到50ml,作为样品溶液。液体中皂素含量由前述的紫外可光光度法确定。
实施例5
实施例5所述皂素提取工艺与实施例4大致相同,不同之处在于:磁性固体酸的添加量为0.4121g,乙醇为10ml
实施例6
实施例6所述皂素提取工艺与实施例4大致相同,不同之处在于:磁性固体酸是实施例4中回收得到的磁性固体酸,质量为0.7071g,氯仿添加量为140ml
硫酸对比例
采用传统硫酸酸法水解黄姜中皂素提取,其具体步骤与实施例5大致相同,不同之处在于:将磁性固体酸替换为含等质量的浓硫酸,对比硫酸水解黄姜皂苷元的效果。各样品检测及计算结果见表1
表1不同应用例和对比例所得样品溶液的吸光值测试结果
注:产物得率以皂苷粗品为原料,计算而得
上述结果表明:本发明将磁性固体酸用于替代无机酸水解黄姜,其提取皂素得率(实施例2和实施例3的平均值)比同质量硫酸水解高约35.9%,且可以重复回收利用,活性依旧很好,且本发明所得新型磁性纳米固体酸催化剂对设备无腐蚀,环境友好,最重要可回收利用,损失率很小,适合推广应用。
以上对本发明的实施例进行了示例性说明,但所述内容仅为本发明的较佳实施例,不能被认为用于限定本发明的实施范围。凡依据本发明申请范围的均等变化与改进等,均应归属于本发明的专利涵盖范围之内。
- 还没有人留言评论。精彩留言会获得点赞!