官能化石墨烯的生产的制作方法
本发明涉及在电化学电池中生产官能化石墨烯以及相关的官能化石墨纳米片结构的方法。
背景技术:
安德烈·盖姆(andregeim)教授和康斯坦丁·诺沃消洛夫(konstantin(kostya)novoselov)教授于2004年在曼彻斯特大学首次报道了石墨烯的分离。由于其非常理想的物理特性和电子特性,石墨烯立即引起了人们的兴趣。
此后,已经使用了各种方法以制备单层和少量层形式的石墨烯。这些方法包括石墨质电极的电化学剥离。例如,在wo2012/120264中,德莱夫(dryfe)和金洛克(kinloch)描述了通过将烷基铵阳离子电化学插入到石墨中生产石墨烯的方法。
在wo2013/132261中,描述了一种用金属离子和有机离子发生对石墨的双重嵌入(doubleintercalation)的方法。另见[abdelkader,2014]。
在wo2015/019093中描述的另一种电化学方法,该电化学方法使用无溶剂离子电解质,其中电解质选自于:(i)离子液体;(ii)低共熔溶剂;和(iii)固态离子导体。
然而,石墨烯在各种应用中的加工性能因其在大多数常见的低沸点溶剂中的不溶性而受到很大阻碍。共价官能化被认为是提高石墨烯溶解度的方法之一。官能化也可用于改变材料的电子特性。
迄今为止,大多数工作集中在预制石墨烯的官能化上。例如,孙(sun)等人报道了热膨胀石墨的化学官能化,然后对其进行超声处理以引发剥离[sun,2010]。该方案显示出提高了石墨烯在n,n’-二甲基甲酰胺中的溶解度。
恩格勒特(englert)等人报道了用4-叔-丁基重氮苯/4-磺酰基苯基重氮氯化物的化学剥离石墨烯的本体官能化(bulkfunctionalisation)[englbert,2011]。该官能化防止了石墨烯聚合并改善了其在氯仿中的溶解度。
钟(zhong)和斯瓦格(swager)报道了一种方法,在该方法中,他们在两步法中首先在碳酸丙烯酯电解质中将li+和四丁铵离子(tba)电化学地嵌入石墨中(嵌入的li+离子被认为在第二步中与较大的tba离子进行离子交换)。然后将得到的膨胀石墨浸入到含有4-溴重氮苯四氟硼酸盐的电解质中并电化学地官能化。然后用剪刀将膨胀的官能化石墨切割并超声处理[zhong,2012]。
技术实现要素:
本发明提供了方便的石墨同时电化学官能化和剥离,以提供边缘官能化的石墨烯。
第一方面,本发明提供了一种在电化学电池中生产官能化石墨烯和/或厚度小于100nm的官能化石墨纳米片结构的方法,其中所述电池包括:
(a)负电极,所述负电极为石墨质的(graphitic);
(b)正电极;以及
(c)电解质,所述电解质为溶剂中的离子且含有重氮物质;
其中所述方法包括使电流通过所述电池,以便:
(i)发生重氮物质的电化学还原,以生成在所述负电极处经受接枝反应的官能化物质;以及
(ii)将离子嵌入至所述负电极中,以发生剥离;
其中,(i)和(ii)同时发生。
所述负电极为保持在两个电极之外的最大负电势处的电极。也可使用参比电极。
换言之,本发明提供了一种在电化学电池中生产官能化石墨烯和/或厚度小于100nm的官能化石墨纳米片结构的方法。
所述重氮物质经受电化学还原、释放氮气和反应性基团,所述反应性基团被接枝到石墨质电极和/或初期的石墨烯和石墨纳米片结构的层。也就是说,所述重氮物质可称为r-n2+,其经受还原成官能化物质r*(自由基物质),逸出氮气。然后r*接枝到碳片材的边缘。术语接枝反应是指取代基与片材的共价结合。在本领域中,该反应也称为“共价修饰”。
发明人观察到,接枝反应主要发生在石墨烯片材的边缘处,导致边缘官能化的石墨烯和/或厚度小于100nm的边缘官能化的石墨质纳米片。与基面官能化相反,该边缘官能化是有利的。
重要的,嵌入(导致剥离)和官能化同时发生。换言之,在通过电池施加一次电势差(电压)的过程中,在单个过程步骤中发生嵌入、剥离和接枝反应。在电极之间,无需消除电势差或确实改变电势差。
类似地,在嵌入、剥离和接枝反应过程中无需在任何时间消除负电极,例如向电池中添加进一步的试剂。
如本文所述,有利地,发明人发现石墨质电极可在单个步骤中被嵌入、剥离并官能化。
这意味着,石墨质负电极(起始材料)可选自天然石墨和合成石墨。在这方面,本发明不同于钟(zhong)和斯瓦格(swagger)描述的方法,后者将官能化引入至膨胀电极,而该膨胀电极已经经历了两次单独的嵌入步骤[swagger,2012]。
换言之,本发明提供了一种在电化学电池中生产官能化石墨烯和/或厚度小于100nm的官能化石墨纳米片结构的方法,其中所述电池包括:(a)负电极,所述负电极为石墨质的;(b)正电极;和(c)电解质,所述电解质为溶剂中的离子并含有重氮物质;其中,所述方法包括使电流通过所述电池,以便:(i)发生重氮物质的电化学还原,以生产在所述负电极处经受接枝反应的官能化物质;以及(ii)将离子嵌入至所述负电极中,以发生剥离。
石墨是指通过嵌入或其他方法不膨胀的材料。换言之,平均层间距离小于0.5nm,例如约0.335nm。
如本文所述,有利地,发明人发现石墨质电极可在单一的施加电势下、单个步骤中被嵌入、剥离并官能化。
因此,在另一方面,本发明提供了一种在电化学电池中生产官能化石墨烯和/或厚度小于100nm的官能化石墨纳米片结构的方法,其中所述电池包括:(a)负电极,所述负电极为石墨质的;(b)正电极;和(c)电解质,所述电解质为溶剂中的离子并含有重氮物质;其中,所述方法包括使电流通过所述电池,以便:
(i)发生重氮物质的电化学还原,以生产在所述负电极处经受接枝反应的官能化物质;以及
(ii)将离子嵌入至所述负电极中,以发生剥离;
其中,(i)和(ii)在单一的施加电势下发生。
在某些情况下,所述负电极为石墨箔。
在某些情况下,所述单一的施加电势差小于|3.0v|。例如,其可以介于-2.5v和-3.0v之间。单一的施加电势施加持续时间,该持续时间可例如介于1小时和6小时之间。在某些情况下,施加的电势在持续时间内变化不超过±1v。
电解质适当地为有机溶液,但在一些实施方案中可以是离子液体。合适的有机溶剂包括但不限于,二甲基亚砜(dmso)、n,n-二甲基甲酰胺(dmf)或n-甲基-2-吡咯烷酮(nmp)。
有趣地,重氮官能化有助于剥离,使得甚至通常太小而不能剥离石墨以生产石墨烯和石墨质纳米片结构的离子、例如锂离子,也可以在不包括其它嵌入物质的情况下使用。
发明人已经证明,反应过程可以使用电解质来执行,所述电解质为锂离子的溶液、铯离子的溶液、或四烷基铵离子的溶液,但是应当理解,本发明不限于这些离子。因此,合适的阳离子包括锂、铯和四烷基铵。
在某些情况下,在电解质中基本上只有一种阳离子(除了溶解在电解质中且通常为阳离子的重氮物质)。在本文中基本上指的是至少90mol%的阳离子,优选地至少95mol%,更优选地至少97mol%。在某些情况下,在电解质中只有一种阳离子(除了重氮物质)。
如本文所述,发明人发现,铯离子是有吸引力的嵌入物质。铯的嵌入密度被认为是类似与锂的嵌入密度,但铯离子的半径与石墨片材之间的间隙距离尺寸匹配良好。有趣地,当铯是电解质中唯一的阳离子而没有任何官能化时,铯可用于剥离石墨。
因此,在某些情况下,嵌入物质为铯离子。铯离子可提供在有机溶剂中。优选地,铯离子提供在有机溶剂中,例如dmso、碳酸烷基酯、dmf或nmp,更优选地dmso、dmf或nmp。
换言之,电解质可以是有机溶剂中的铯离子的溶液(例如,诸如csclo4的铯盐的溶液)。
另一方面,本发明提供了一种在电化学电池中生产石墨烯和/或厚度小于100nm的石墨纳米片结构的方法,其中所述电池包括:
(a)负电极,所述负电极为石墨质的;
(b)正电极,所述正电极可以是石墨或其他材料;以及
(c)电解质,所述电解质为包含铯离子的溶剂中的离子;
且其中,所述方法包括使电流通过所述电池的步骤。
适当地,所述负电极选自高度有序的热解石墨、天然石墨和合成石墨。
在某些情况下,所述方法在20℃至100℃的温度下实施。优选地,所述方法在低于50℃下、例如在室温下实施。
在某些情况下,所述石墨烯或厚度小于100nm的石墨纳米片结构通过至少一项技术从所述电解质中分离,所述至少一项技术选自:
(a)过滤;
(b)使用离心力以沉淀石墨烯或石墨纳米片结构;以及
(c)在两种互不相溶的溶剂的界面处收集石墨烯或石墨纳米片结构。
如本文例示,铯离子可被提供为csclo4的有机溶液。例如,铯离子可被提供为dmso中的溶液。
具体实施方式
现将参照附图讨论说明本发明的原理的实施方案和实验。
附图
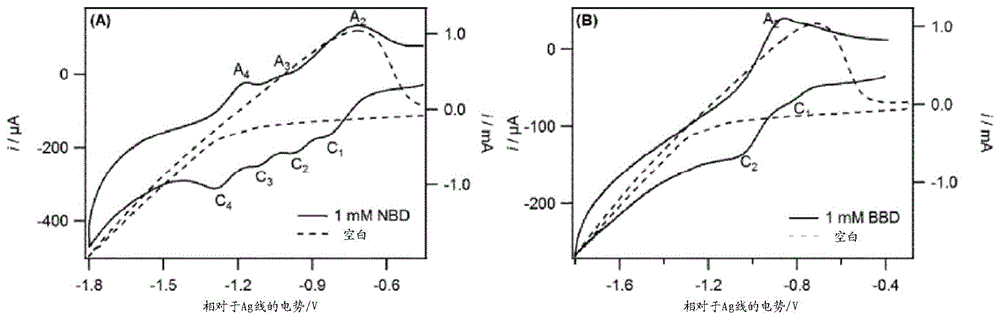
图1示出了在含有(a)1mmnbd和(b)1mmbbd的n2气氛下,在0.1mcsclo4的dmso中在hocg电极处记录的循环伏安图。在各情况下,电势从-0.3v的初始电势以100mvs-1在-1.8和-0.3v之间扫描。在各情况下,虚线示出了在没有重氮物质的情况下获得的数据且对应于右轴,而实线示出了在有重氮物质的情况下获得的数据且对应于左轴。
图2示出了代表硝基重氮苯物质(nitrobenzenediazonium,nbd)阳离子在石墨电极处的电化学还原,以及随后的接枝步骤和还原步骤的反应。
图3示出了在0.3mcsclo4的dmso中含有40mm溴重氮苯(bromobenzenediazonium,bbd)的溶液中,在指定时间向等压成型的石墨工作电极施加-4.0v后记录的紫外可见光谱(uv-visiblespectra)。
图4示出了具有不同浓度的(a)bbd(底部至顶部为石墨质的、电化学剥离的石墨烯,20mm、40mm和100mm)和(b)nbd(底部至顶部为石墨质的、电化学剥离的石墨烯,1mm、40mm和100mm)的原位电化学剥离石墨烯片材和官能化的石墨烯片材的代表性拉曼光谱。
图5示出了(a)电化学剥离的石墨烯、g-nbd和g-bbd的宽扫描xp光谱;(b)在n1s区域的g-nbd的高分辨率xp光谱以及(c)在b3p区域的g-bbd的高分辨率xp光谱。所有的峰位通过将c1s信号的结合能设定为285ev来进行电荷校正。
图6示出了在40mmnbd和0.3m的csclo4的dmso中,相对于ag线,在-4.0v处通过石墨的电化学剥离获得的g-nbd的sem图像:(a)沉积在si/sio2晶片上的dmso稀释的分散体;和(b)沉积在si/sio2晶片上重新堆叠的薄膜。
图7显示了(a)电化学剥离的石墨烯薄片的tem图像、(b)40mmg-nbd的tem图像、(c)电化学剥离的石墨烯的afm图像、以及(d)40mmg-nbd的afm图像。
图8示出了(a)分散在指定溶剂的100mmg-nbd的典型的紫外光-可见光吸收光谱;(b)水/ipa(体积比为1:1)中的g-nbd分散体的典型的紫外光-可见光吸收光谱;(c)分散在指定溶剂的100mmgbbd的典型的紫外光-可见光吸收光谱;以及(d)水/ipa(体积比为1:1)中的gbbd分散体的紫外可见吸收光谱。在各情况下,在测量前将官能化石墨烯稀释五倍。
图9示出了(a)使用由指定样品构造的对称纽扣电池以100mvs-1在6.0mkoh(aq)中记录的循环伏安图,电压在0.0v至1.0v之间扫描;(b)在40mmg-nbd纽扣电池中在0.5v(初始点位)和1.3v之间以(从上到下)100、85、60、45和20mvs-1在6.0mkoh中获得的循环伏安图;(c)在指定电极处以0.5ag-1获得的充-放电曲线。
图10示出了相比于石墨(虚线)的g-aqd(实线)的代表性拉曼光谱。
图11比较了石墨烯(内曲线)和aqd(外曲线)的循环伏安图。
图12示出了在含有15mmaqd的n2气氛下,以100mvs-1在0.1mcsclo4的dmso中hopg电极处记录的循环伏安图。电势从0.2v的初始电势在-1.8v和0.2v之间扫描。
图13示出了g-aqd的示意性视图。
重氮反应
石墨烯的重氮官能化由于其多功能性、反应简单性和对sp2杂化碳中心的高反应性而具有吸引力。重氮物质在与富电子表面相互作用时容易产生自由基,例如芳基。然后自由基快速地与sp2杂化的碳原子反应。
以下方案说明了硝基重氮苯物质(nbd)的反应步骤。
应当理解的是,如本文所用,术语“重氮物质”是指带有-n2基团的分子实体,典型地表示为-n+≡n,尽管可以表示其他互变异构形式(例如=n+=n--)。重氮物质适当地提供为盐,其可称为重氮盐,该盐包括重氮物质和反离子。
应当理解的是,可以使用其他合适的重氮物质,其不一定是苯基的。可以使用任何合适的r-n2+物质。例如,r可以是任意取代的烷基、烯基、芳基或杂芳基。
适合地,r为任意取代的芳基或杂芳基。芳基可以指c6-20环系,例如,诸如苯基和萘基的c6-10环系。芳基包括醌基团,例如蒽醌。杂芳基可以指含有一个或多个杂原子的c5-20环系,例如,含有一个或多个杂原子的c5-6环系。
例如,r可以是可选地取代的苯基。
例如,r可以是可选地取代的蒽醌,例如,r可以是未被取代的蒽醌。
换言之,r-n2+可以是蒽醌-1-重氮,也称为aqd。
可选的取代基可包括卤素(f、cl、br、i)、oh、no2、cn、c1-6烷基(例如me)、c2-6链烯基、c1-6卤代烷基(例如cf3、ccl3)、cooh、so3h。优选的可选的取代基可包括cl、br、no2、cn、cf3、ccl3和so3h。
可选地,官能团可经受随后的反应/衍生以提供其他序列。该方法利用第一原位官能化步骤来(主要地)生产边缘官能化石墨烯,随后可从该边缘官能化石墨烯中添加更复杂的序列。
应当理解,取代基可增加重氮物质的稳定性和/或提供用于提高石墨烯的加工性能的官能度和/或提供用于进一步反应的操作。例如,在某些情况下,取代基为卤素或no2,例如本文中示例性的br或no2。
应当理解,重氮物质在0℃下存储和操作是适当地稳定的,更优选地,重氮物质在室温下操作是稳定的。
在某些情况下,重氮物质选自于:
重氮物质的反离子可以是任意合适的阳离子,例如卤化物或硼酸盐物质。本领域中已知,某些重氮物质可以作为四氟硼酸盐分离。这些盐通常显示出期望的稳定性。因此,优选的重氮物质用作四氟硼酸盐。
在一些实施方案中,重氮物质为4-硝基重氮苯(nbd)。例如,可使用4-硝基重氮苯四氟硼酸盐。
在一些实施方案中,重氮物质为溴重氮苯(bbd)。例如,可使用4-溴重氮苯四氟硼酸盐。
在一些实施方案中,重氮物质为蒽醌-1-重氮(aqd)。例如,可使用蒽醌-1-重氮氯化物。
优先的边缘官能化
发明人观察到,该过程对边缘官能化为高度选择性的,且如果有的话,在获得的石墨烯片材的基面上引入很少的官能化。
不希望受到任何特定理论的约束,发明人首先将其归因于由片材中的电子/电荷分布导致的边缘官能化的热力学偏好,其次将其归因于官能化的发生是在大块材料的剥离开始之前进行的事实。随着石墨烯片材(已经边缘官能化的)从阴极产生,官能化继续在电极材料处优选。
因此,主要边缘官能化的材料在不牺牲与大部分无缺陷的石墨烯片材相关的期望的电子特性和物理特性的情况下的提供了改善的溶解性和加工性能的优点。
重要的是,发明人观察到,与可预期的相反,将取代基接枝到各个片材的边缘不会显著地影响离子和其他物质的嵌入以引起/帮助剥离。不希望受任何特定理论的束缚,发明人推测边缘官能化可能确实帮助石墨的各个层的分离,引起有限程度的膨胀,而该膨胀甚至可以帮助阳离子物质的嵌入。
例如,在使用nbd和cs+的剥离和官能化的情况下,发明人观察到与在空白电解质中使用cs+的剥离相比,过电势仅增加0.3v,没有明显的嵌入损失。
实际上,发明人认为层的边缘的官能化也可通过在电极处生成氮气(反应的副产物)来帮助剥离过程。
例如,尽管已经证明单独的cs+在电化学电池中剥离石墨烯,但是发明人认为,逸出的气体可帮助片材从电极上分离。该气体可进入片材之间的间隙空间,片材之间的范德华力已经因嵌入物质的存在而削弱且有助于驱动片材分开。氮气的逸出又引起电极附近的浊度,帮助剥离的片材或部分剥离的片材的脱落。
有趣的是,发明人已经观察到,边缘官能化的帮助提供了甚至允许使用非常小的嵌入物质。例如,li+能够嵌入石墨中的层之间,但不会引起剥离以生产石墨烯。这归因于li+的小尺寸(仅0.146nm)。因此使用另外的、更大的阳离子,例如四甲基铵阳离子[abdelkader,2014]。
然而,发明人已经观察到,如本文描述的重氮物质与li+的组合导致石墨质电极的官能化和剥离,产生官能化的少量层石墨烯。不希望受任何特定理论的束缚,发明人将其归因于:
(i)官能化有助于撬开层的边缘,以及
(ii)嵌入的li+的存在削弱了间隙力,
因此,由逸出的氮气施加的力足以实现层的分离。
重要的是,某种程度的嵌入似乎是必要的。在对照实验中,发明人用nbd而无金属离子(或其他嵌入离子,例如铵物质)来进行电化学反应。尽管随着官能化的发生显著的气体逸出,但未观察到剥离。
负电极
负电极为石墨质的。天然的石墨和合成的石墨均可使用。在某些情况下,该电极为天然石墨。例如,该电极可以是石墨箔或石墨棒。在某些情况下,该电极为合成石墨。
在某些情况下,电极为等压成型石墨。等压成型石墨也称为各向同性石墨、等静压石墨以及等静压压制石墨。由于其均匀性,可优选等压成型石墨。
通常,在本文中描述的方法中可使用任何固体石墨电极。在一些实施方案中,负电极可以是浇包设计(ladledesign)以避免电极分解成大块的问题。在另一实施方案中,石墨粉末保持在诸如平纹棉布的多孔织物中,或诸如镍网的导电网中。在其他实施方案中,石墨负电极可被保持在液-液界面处。在这样的实施方案中,负电极可以是其上放置有石墨薄片的液态金属,例如汞或镓,,以允许当石墨材料剥离成期望的材料时,与石墨材料持续接触。
正电极
正电极可由本领域技术人员已知的任何合适的材料组成,因其除了为阴离子提供对电极外,在石墨烯的生产中并不起作用。优选地,正电极由惰性材料、例如金、铂或碳制成。
当在正电极处的反应产生气体时,该电极的表面积尽可能地大以防止气泡润湿该电极和/或破坏在负电极上的过程。正电极和/或参比电极也可放置于薄膜或分子筛中,以防止电解质中或任一电极处的不期望的反应。
在一实施方案中,两个电极可适当地由石墨制成,且在两个电极间切换的电势依次在每个电极上实现剥离和官能化。
电池电势和电流密度
电池的工作电势将至少为还原嵌入的标准电势。可使用超电势,以便增加反应速率并驱动阳离子进入到负电极处的石墨的通道。优选地,相对本领域技术人员已知的合适的参比,使用1mv至10v的超电势,更优选地为1mv至5v,更优选地为1v至5v。在某些情况下,超电势约为4v。在仅具有两个端子且没有参比的电池中,可以在电极上施加更高的电势,但是大量的电势降将发生在电池电阻上,而不是在电极处起到超电势的作用。在这些情况下,施加的电势可以达20v或30v。
有利地,发明人已经发现官能化和剥离都可以在单一的电势下、在单个步骤中有效地实现。因此,在一些情况下,在施加电流期间,超电势的变化小于±1v,例如小于±0.5v。
负电极处的电流密度可通过电极的表面积和使用的超电势的组合来控制。
电解质
电解质适宜地为有机溶剂中的离子,但在某些实施方案中可以是离子液体。
可使用的溶剂包括(n-甲基-2-吡咯烷酮)nmp、烷基碳酸盐(如碳酸丙烯酯)、dmso(二甲基亚砜)、dmf(n,n'-二甲基甲酰胺)及它们的混合物。在一个实施方案中,使用的溶剂对石墨烯或石墨纳米片结构具有亲和力,使得在电极处生成的材料被溶剂带走。在另一个实施方案中,溶剂对石墨烯或石墨纳米片结构没有亲和力,以便生成的材料落到电化学电池的底部,允许容易地收集生成的石墨烯。
其他方法步骤
由本发明的方法产生的官能化的石墨烯或厚度小于100nm的石墨纳米片结构可通过多种分离技术从电解质中分离出来,所述分离技术包括:
(a)过滤;
(b)使用离心力以沉淀石墨烯或石墨纳米片结构;以及
(c)在两种互不相溶的溶剂的界面处收集石墨烯或石墨纳米片结构。
电化学剥离的石墨烯或石墨纳米片结构在剥离之后可以被进一步处理。例如,所述材料可以使用超声能量和本领域技术人员已知的其他技术来进一步剥离,以降低薄片尺寸和/或石墨烯层数。
操作温度
电池在允许生产所期望的材料的温度下被操作。
电池可在至少10℃的温度下被操作,优选至少20℃。最大电池操作温度可以是100℃,且更优选90℃、80℃、70℃或50℃。在一些实施方案中,电池可在至少30、40或50℃的温度下被操作。最大电池操作温度可以高达120℃。最佳的操作温度将随着溶剂的性质变化。在本发明中,至高可以在电解质的沸点下操作电池。
石墨烯和石墨纳米片结构
在本申请中,术语“石墨烯”用于描述理想地由一至十个石墨烯层组成的材料,优选地,其中产品中层的数量的分布是可控的。该方法也可用于制备厚度在100nm以下的石墨纳米片结构,更优选地厚度在50nm以下,更优选地厚度在20nm以下,且更优选地厚度在10nm以下。取决于期望的形态,成产的石墨烯薄片的尺寸可从纳米变化到毫米变化。
在实施方案中,生产的材料为具有至多十层的石墨烯。生产的石墨烯可具有一层、二层、三层、四层、五层、六层、七层、八层、九层或十层。可优选地,生产的材料基本上不含氧化石墨烯。“基本上不含”意味着按重量计小于10%的氧化石墨烯,优选地按重量计小于5%,更优选按重量计小于1%。
在实施方案中,生产的材料可包括按重量计至少10%的具有至多十层的石墨烯,优选地按重量计至少25%、更优选地按重量计至少50%的具有最大至十层的石墨烯。
本发明的方法生产石墨烯和/或厚度小于100nm的石墨纳米片结构。在实施方案中,本方法生产石墨烯或厚度小于100nm的石墨纳米片结构。在实施方案中,本方法生产石墨烯和厚度小于100nm的石墨纳米片结构。在实施方案中,本发明的方法生产石墨烯。在实施方案中,本方法生产厚度小于100nm的石墨纳米片结构。例如,本发明的方法可生产石墨烯、或石墨烯和厚度小于100nm的石墨纳米片结构的组合。
在实施方案中,按表面积计,本方法生产的石墨烯比厚度小于100nm的石墨纳米片结构更多,优选地,其中按表面积计,基本上通过该方法生产的所有的材料都是石墨烯(其中,按表面积计,通过该方法生产的材料中至少90%、优选地至少95%、更优选地至少98%、例如至少99%为石墨烯),例如,其中通过该方法生产的所有材料都是石墨烯。在实施方案中,按重量计,该方法生产的石墨烯比厚度小于100nm的石墨纳米片结构更多,优选地,其中,按重量计,通过该方法生产的基本上所有的材料都是石墨烯(其中,按重量计,通过该过程生产的材料的至少90%、优选地至少95%、更优选地至少98%、例如至少99%为石墨烯),例如,其中由该过程生产的所有材料都是石墨烯。因此,在一些实施方案中,石墨烯由一至五个石墨烯层组成,优选地一至四个石墨烯层、更优选地一至三个石墨烯层,例如一至两个石墨烯层、如一层。因此,生产的石墨烯可具有一个、二个、三个、四个、五个、六个、七个、八个、九个或十个层。
实施例
以下实施例,包括进行的实验和得到的结果是出于说明的目的而提供,且不旨在限制本发明。
材料和试剂
4-硝基重氮苯四氟硼酸盐(97%)、4-溴重氮苯四氟硼酸盐(96%)和无水二甲基亚砜(99.9%)从西格玛奥德里奇(sigma-aldrich)获得。蒽醌重氮氯化物从美国bocsciences获得。高氯酸铯(99%)从飞世尔科技(fisherscientific)获得。所有的电化学测量使用电化学工作站恒电势仪模型pgstat302n(万通电化学工作站(metrohmautolab),荷兰)来完成。等压成型石墨(>99.95%)棒从石墨商店(graphitestore)购买,且石墨箔(99.8%)从阿法埃莎(alfaaesar)获得。聚四氟乙烯从具有0.1μm孔径的omnipore膜过滤器(jvwp01300)获得。密理博水(milliporewater)(18.2mωcm)从密理博超纯水净化系统(milli-qwaterpurificationsystem)获得。高度取向的热解石墨(hopg)zyb质量从美科精微(micromechanicsltd)(香港)购买。
cs+和重氮盐的电化学
新切割的高度取向的热解石墨工作电极、pt网状对电极和ag线参比电极用于电化学测量。ag线的电势在几mv内稳定超过4小时。在进行循环伏安法之前,将n2气体鼓入电解质中30分钟,并且在电化学测量期间,在电解质上方保持n2气氛。电解质由无水二甲基亚砜(dmso)中的0.1mcsclo4和1mm4-硝基重氮苯四氟硼酸盐(nbd)或dmso中的0.1mcsclo4和1mm4-溴重氮苯四氟硼酸盐(bbd)组成。
电化学剥离和官能化
石墨烯的电化学剥离和官能化使用三个电极装置执行,该三个电极装置由等压成型石墨棒/石墨箔工作电极、银线参比电极和等压成型的石墨棒对电极组成。暴露于电解质的工作电极的有效面积约为12cm2。
电解质通过将0.3mcsclo4和各种浓度(1mm、40mm和100mm)的nbd或bbd溶解在无水dmso中来制备。同时的电化学剥离和官能化使用计时电流法来执行,通过在连续搅拌下相对于ag线施加-4.0v的电势2小时。以类似的方式,非官能化石墨烯在溶液中在相同的电势处被剥离,该溶液在二甲基亚砜中仅含有0.3mcsclo4。然后被剥离的产品用大量丙酮和超纯水来洗涤,并在60℃下真空干燥过夜。官能化粉末通过超声处理30分钟分散在期望的溶剂(水、异丙醇、以及水与异丙醇的混合物)中。得到的混合物在4000rpm下离心30分钟,上清液通过使用移液管提取而不扰动残余物。
为了验证实验装置独立于阳离子,使用0.3m的四乙基四氟硼酸铵代替csclo4,并得到了类似的结果。
实验装置还用40mmnbd和0.3mliclo4来重复。再一次,获得了类似的结果。
剥离产物的特征
拉曼光谱通过使用拉曼光谱仪显微镜(renishawinviamicroscope)来获得,该拉曼光谱仪显微镜具有532nm的激发激光,该激发激光在1mw的低功率下操作,光栅为1800l/mm,100×物镜。用于拉曼测试的样品通过将石墨烯的分散体滴涂在si/sio2晶片上来制备,并在100℃下在电热板上干燥以蒸发溶剂。使用在15kv下操作的xl30fei环境扫描电子显微镜来进行扫描电子显微镜(sem)分析,且样品通过将石墨烯的分散体滴涂在si/sio2晶片上来制备。用于afm测量的样品通过在si/sio2上喷涂石墨烯的分散体来制备。afm模型为且afm在环境条件下以轻敲模式操作。透射电子显微镜(tem)图像通过使用日本电子实验室(jeol)2000fxtem、在200kv下操作来记录。x射线光电子能谱(xps)使用奎托斯(kratos)axisultradld光谱仪来进行,该光谱仪具有单色的alkαx射线源(e=1486.6ev,10ma发射)、半球形电子能量分析仪以及多通道板和延迟线检测器(dld)。石墨烯的分散体浓度使用紫外光-可见光光谱、通过使用dh-2000-bal模型(海洋光学)来测量。官能化石墨烯的消光系数通过科尔曼(coleman)等人描述的方法来确定[hernandez,2008]。
用于超级电容器测试的膜制备
非官能化石墨烯、nbd-官能化石墨烯或bbd-官能化石墨烯的膜通过使用注射泵分配器(新时代泵系统公司,纽约(newerapumpsystems,inc,ny))在聚四氟乙烯(ptfe)上过滤已知体积的分散体来制备。然后,将膜在80℃的烘箱中干燥过夜。纽扣电池组件在具有对称活性材料的标准cr2032纽扣电池硬件中制备。电池通过将两个对称膜背靠背堆叠来组装,活性材料接触集电器[bissett,2015]。在使用液压卷边机(msk-160d)密封纽扣电池之前,加入几滴脱氧的6mkoh(水溶液)以填充电极。比电容使用由斯托勒(stoller)和劳夫(ruoff)建立的最佳实践方法来计算[stoller,2010]。
重氮盐的电化学
发明人检查了附着在石墨上的重氮物质是否阻碍了cs+的嵌入。先前已经观察到,用重氮物质接枝碳电极使表面钝化,并且通常接枝的表面趋于对简单的氧化还原介体、如二茂铁和铁氰化物无活性[参见,例如saby,1997]。
图1a显示了在具有nbd和没有nbd的hopg处在n2气氛下记录的循环伏安图(cvs)。在空白电解质(dmso中的0.1mcsclo4)中,当电极的电势在负方向上扫描时,阴极电流在约-1.3v处开始流动,并在-3.5v处观察到宽峰,这是由于cs+的嵌入。当电极电势进一步扫描为-4.0v的负电压时,由于dmso的还原,看到电流急剧增加。
相反,含有nbd的cv响应显示出一系列氧化和还原过程(图1b)。沿负方向扫描电极电势产生了四个还原峰。削峰实验显示c2、c3和c4分别与a2、a3和a4相关。c1被发现是不可逆的,这表明电生成的产物不稳定且与表面反应。根据等式1(图2),c1归因于硝基苯自由基的形成,并且已知这种自由基对碳基电极具有高反应性。参见,例如,[allongue,1997]。
c2和c3分别归因于硝基自由基和二价阴离子物质的形成(图2的等式2和3)。硝基苯在单电子转移过程中可逆地还原以产生稳定的自由基阴离子,但是二价阴离子物质在弱酸存在下易于快速的质子化。由于cv中a3与c3的峰值电流比小于0.5,因此二价阴离子可通过从痕量水或dmso本身中接受质子以质子化形成不稳定的物质(等式4)。巴德(bard)及同事报道了,二价阴离子在弱酸(异丙醇)存在下,在损失羟基离子后可分解为亚硝基苯[smith,1975]。康普顿(compton)及同事也报道了在质子惰性的室温离子液体中二价阴离子物质的不稳定性[silvester,2006]。
二价阴离子分解后,形成的亚硝基苯可通过质子惰性的电解质中的双电子和双质子转移过程还原为苯基羟胺,并可通过水性系统中的四电子和四质子转移过程还原为氨基苯。由c4测量的电流约为由c2或c3测量的电流的两倍,这表明c4是双电子转移过程。因此,发明人将c4归因于如等式5所示的苯基羟胺的形成。
可能从cv中最重要的观察结果是通过硝基苯物质对hopg表面的接枝不会阻碍cs的嵌入。相比于空白电解质中的cs+嵌入,其超电势增加0.3v。因此,发明人证明了单个步骤中的官能化和剥离的可能性。
4-溴重氮苯(bbd)的电化学与nbd的电化学不同之处在于仅观察到两个还原峰和一个氧化峰(参见图1b)。如先前讨论,第一还原峰(c1)是由于溴苯自由基的形成,其与hopg表面快速地反应。c2与a2有关,且c2可能由于可逆的单电子转移以形成自由基物质。cs+的嵌入也发生在与空白电势相同的电势处,这表明在hopg表面上溴苯的存在不会改变用于cs+的插入的电势。
电化学剥离期间的重氮反应过程
根据对cs+和重氮电化学的理解,石墨棒或石墨箔用于在单一的施加电势下同时的剥离和官能化。选择相对于ag的-4.0v作为电势,因为在该电势处,cs+嵌入在扩散可控的速率下发生,且在该电势处,重氮物质的还原也以非常温和的速率发生。
cs+的离子尺寸为0.338nm,其类似于石墨的层间距(0.335nm)。此外,溶剂化的cs+的尺寸预计高于石墨的层间距。事实上,在dmso的溶剂化的cs+中,cs-o键长为0.306nm,且各cs+与八个dmso分子形成溶剂化。
在电解期间,在电极表面处检测到了显著的气体逸出。气体逸出伴随着电解几分钟内的石墨的剥离。
在剥离期间通过紫外-可见光(uv-vis)光谱监测重氮反应的速率。图3示出了在含有0.3mcsclo4和40mmbbd的溶液中bbd的反应历程,该bbd的反应历程作为电解时间(t)的函数。在287nm处观察到宽的吸收峰,这是由于在t=0时来自重氮的电子跃迁[mu,2004]。随着t增加至10分钟,该峰的强度逐渐降低,并且在t=30分钟时迅速降低,这表明85%的重氮已经在电解30分钟内被反应。在t=2小时处实现了完全的重氮反应。此外,在t>30分钟时在256nm处出现新的吸收峰,并且还注意到在可见光范围内的适度的恒定吸收。这种新的吸收峰可能是由于剥离的官能化石墨烯材料的电子跃迁。因此,所有的原位电化学剥离和官能化都进行了2小时。
剥离产物的表征
拉曼光谱用于确认少量层官能化的石墨烯的形成,且图4示出了不同的重氮浓度下官能化的石墨烯的拉曼光谱。为了对比,还提供了不存在重氮时使用cs+电化学剥离的石墨烯(在图中标记为eeg),并与起始石墨材料的拉曼光谱进行对比。石墨的拉曼光谱示出了在1579cm-1和2719cm-1处的两个强峰,该两个强峰分别对应g带和2d带。g带是由于sp2杂化碳的e2g振动模式,2d带是由具有相反波矢的两个声子的散射引起的二阶振动。由于d带,在1349cm-1处看到另外的小峰,且当石墨材料中存在缺陷时,该带为活性的。晶格缺陷可以由通过共价化学形成的sp3杂化或由sp2-共轭碳中的物理缺陷或边缘而引起。
在不存在重氮时电化学剥离后,2d带和g带的峰位分别移位约33cm-1和8cm-1。此外,2d峰的形状从石墨的典型的宽的非对称形状改变为对称的线形。这表明少量层石墨烯片材的形成。
对于在存在重氮盐的情况下剥离的样品,相对于电化学剥离的石墨烯,注意到d带的强度的显著增加。发现d带的强度强烈依赖于重氮盐的浓度。
d带的演变伴随着d′带在1614cm-1处的强度的增强。电化学剥离的石墨烯中d带与g带的强度比(id/ig)为0.28,且当100mmnbd和100mmbbd用于原位官能化时,d带与g带的强度比值分别增加至2和3。广泛接受的是,d带与g带的强度比是石墨烯薄片中的无序或缺陷的程度。其也用于表征重氮物质的共价官能化的程度[niyogi,2010]。
格林伍德(greenwood)和同事电化学官能化具有一系列重氮盐浓度的原始石墨烯,且他们报道id/ig介于0.1和3.1之间[greenwood,2015]。由于氮和溴的吸电子(p-型掺杂)性质,2d带的强度也降低并变宽。随着重氮浓度的增加g带和2d带的峰位置向上移动,p型掺杂更明显,这一观察结果与报道的文献[niyogi,2010]、[solis-fernandez,2015]、[lim,2010]一致。
如先前的工作报道表面吸附的非共价反应的重氮分子通常出现在1400和1440cm-1之间,拉曼光谱也没有示出与任何物理吸附的重氮相关的谱带。这表明不存在任何表面吸附的非共价反应的重氮分子,并且电化学官能化仅通过共价键发生。
x射线光电子能谱(xps)还用于确认石墨烯的电化学官能化以及评价其化学组成。图5显示了电化学剥离的石墨烯、用硝基重氮苯(g-nbd)官能化的石墨烯以及用溴重氮苯(g-bbd)官能化的石墨烯的宽扫描光谱。在每种情况下,观察到了c1s和o1s的信号,且所有的峰位通过将c1s信号的结合能设定为285ev来进行电荷校正。在全谱扫描中,g-nbd中n1s的存在和g-bbd中br3p的存在证明了用期望的苯基基团官能化石墨烯的成功。此外,在br的原子浓度从1mmbbd中的0.4%增加至100mmbbd中的5.2%的情况下,n的原子浓度从1mmnbd中的0.5%增加至100mmnbd中的4.8%。
高分辨率n1s信号仅在1mmnbd中在399.3ev处显示一个峰,且先前的文献将该结合能位置归因于nh2基团[mendes,2003]。尽管我们的电化学分析表明nhoh最可能是-1.5v处的表面基团,但当剥离和官能化在-4.0v处进行时,nhoh基团可能会经受进一步还原为nh2基团。然而,当nbd浓度增加到40mm及以上时,在no2组中,在氮的典型的结合能处出现了位于405.6ev处的小肩峰。no2基团的演化取决于nbd的浓度。在40mmnbd中,no2基团占19%,且在100mmnbd中其增加至29%。在研究的所有样品中在明显的浓度(>70%)中nh2基团的存在支持了从nbd的电化学分析中得到的结论。
然而,应当注意,no2基团的存在可能表明在电化学剥离后通过自发的原位化学反应在溶液中也发生了一些重氮官能化。
图6a显示了官能化石墨烯薄片(40mmg-nbd)的代表性sem图像。200个薄片的横向尺寸测量表明,薄片尺寸在0.5μm至3.5μm之间变化,并且大部分薄片大约为1μm。如sem、tem和afm图像所示,官能化石墨烯和非官能化石墨烯的形态之间存在明显差异。官能化的石墨烯薄片在其整个表面上显示出强烈的褶皱和波纹的网状,而非官能化的石墨烯薄片呈现折叠的平整表面(参见图6b和7)。对于官能化石墨烯,这样的观察结果先前也注意到了,并归因于由强烈官能化产生的应变。石墨烯片材上的纳米级褶皱减少了单个片材之间的重新堆叠,提供了快速的离子扩散通道并为催化反应提供了更多的活性位点,并且对诸如超级电容器的能量存储装置是有吸引力的。该特征还在聚合物复合材料中提供改善的粘合性和更好的互锁特性。
使用afm测量的非官能化石墨烯的薄片厚度在0.67nm至5nm之间变化,这表明单层石墨烯形成多层石墨烯。因为大量的起皱和官能化,对于官能化石墨烯,其厚度的波动范围在1nm至12nm。
官能化石墨烯的可分散性
石墨烯在溶剂中的分散性取决于石墨烯的表面能和溶剂的表面能之间的匹配,而其长期稳定性由溶剂通过静电排斥或空间位阻稳定石墨烯片材的能力决定。具有与石墨烯相似的表面能的溶剂被认为是分散石墨烯的最有效的溶剂,因为这些溶剂和石墨烯之间的界面张力是最小的。例如n-甲基吡咯烷酮和二甲基甲酰胺的机溶剂已被发现是分散石墨烯的最佳溶剂。
然而,这些溶剂对于多种器官是有毒的,这限制了它们的吸引力。此外,它们的高沸点对薄片的沉积和复合材料的形成造成了问题。诸如氯仿和异丙醇的低沸点溶剂用于石墨烯分散和剥离,但它们具有差的分散稳定性和剥离质量。
在这个方面,改善的水中分散性和低毒性的有机溶剂对于商业应用是至关重要的。石墨烯由于其强的疏水性在水中的溶解度是非常低的,而氧化石墨烯由于其表面官能团而在水中分散。
发明人调查了如本文所述的官能化石墨烯在水和异丙醇(ipa)混合物中的分散性,并将其分散性与未官能化的石墨烯的分散性进行比较。应注意的是,各样品有意地以8000rpm离心30分钟以观察分散体的稳定性,并通过紫外-可见光光谱分析上清液。广泛接受的是,石墨烯的浓度可以通过取660nm处的吸光度值(a660),使用比尔-朗伯定律(beer-lambertlaw)来估算。图8示出了水、ipa和水-ipa混合物中的g-nbd和g-bbd的分散体获得的紫外-可见光光谱。当分散在纯水或ipa中时,g-nbd和g-bbd均显示出了低的a660值。然而,含有ipa和水的混合物以1:1体积比给出了每个样品的a660的最高值,这表明该溶剂混合物对于分散g-nbd和g-bbd是最佳的。水-乙醇混合物用于石墨的剥离,并且在这种情况下,发现石墨烯的分散在混合物中比任何单独的溶剂更高。增强的分散归因于汉森溶解度参数(hansensolubilityparameters,hsp)的改变。
ipa-水混合物中最佳的异丙醇质量分数被报道为石墨烯的55%,这非常接近于该工作中使用的ipa(~60%)的质量分数。
图8a和8c示出了,与nbd官能化石墨烯相比,增加分散溶液中的水浓度对bbd官能化石墨烯的影响更大,这表明溴化石墨烯比氮化石墨烯更具极性。发现g-bbd的分散强烈依赖于bbd的官能化浓度(图8d),这显示出当bbd浓度从1mm增加至100mm时溶解度的增加。相比之下,发现g-nbd的溶解度不依赖于nbd的浓度:发明人观察到,与在100mmbbd中获得的分散体相比,仅用1mmnbd可以实现更高的分散体浓度(图8c)。
g-nbd和g-bbd分散体的吸收系数(α)使用ipa/水混合物以1:1体积比、按照科尔曼(coleman)等人给出的方法[hernandez,2008]来确定。由于吸光度随着浓度的增加而线性增加,因此各分散体均遵循比尔-朗伯定律,并且获得g-nbd的α值为2978±125ml·mg-1m-1和2853±268ml·mg-1m-1。先前在文献中报道了一系列α值。科尔曼等人报道了溶液剥离的石墨烯的分散体在不同溶剂中的值为2460ml·mg-1m-1[hernandez,2008]。洛塔(lotya)等人报道了由表面活性剂稳定的石墨烯的分散体的值为6600mg-1m-1[lotya2010]。康尼斯(konios)等人报道了氧化石墨烯分散体在水中的值为3592mlmg-1m-1[konios,2014]。
使用获得的值来确定g-nbd和g-bbd的溶解度。在ipa/h2o中,与g-bbd的150μgml-1和电化学剥离的石墨烯仅5μgml-1相比,g-nbd显示出了最高的溶解度(250μgml-1)。
这与报道的工作相比是有利的以增强石墨烯溶解度。金(kim)等人报道了石墨烯在高温下在水中的剥离和分散,并获得了6.5μgml-1的最高溶解度[kim,2015],这与我们使用电化学剥离的石墨烯获得的值非常接近,而伍(wu)等人报道了在乙醇和水混合物中为50μgml-1。
显然,如本文所述的具有苯基基团的石墨烯的表面官能化使石墨烯的溶解度提高了两个数量级以上。
官能化石墨烯的电容
如本文所述的生产的官能化石墨烯的电容使用对称纽扣电池结构(cr2032)通过使用循环伏安法和计时电位法来研究。电极通过在预先称重的聚四氟乙烯(ptfe)膜上过滤已知体积的分散体来制备,其中ptfe充当电极和电荷分离器。各电极的典型的质量负载约为0.5mgcm-2。图9a比较了电化学剥离的石墨烯形成的电极和g-nbd形成的电极在脱氧6mkoh(aq)中在0.1vs-1下获得的cv。使用电化学剥离的石墨烯电极获得的cv显示出具有矩形形状的典型的电容行为,并且当扫描电压高达0.9v时没有观察到显著的法拉第反应。相反,随着nbd的官能化浓度从1mm增加至40mm,出现了0.3v(ox1)和0.61v(o2)处的两个明确定义的瞬态氧化峰(ep),以及0.2v(r1)和0.54v(r2)处的两个相应的还原峰。对于每个氧化还原对,阳极峰值电流ip,a与阴极峰值电流ip,c的比值约为1:1。此外,对每个氧化还原反应,ip,a的曲线随着的扫描速率的增加而线性增加,这证明了氧化还原反应是表面受限的过程(图9b)。
xps证明了g-nbd主要含有基于苯胺的基团,并且两个氧化还原峰可归因于苯胺的电氧化。以前的报告示出了苯胺的电化学氧化可产生各种氧化还原对,这些氧化还原对归因于氮宾阳离子、对氨基二苯胺、联苯胺的形成以及聚苯胺膜的降解。当从电化学剥离的石墨烯移动到g-nbd时,在充-放电曲线中法拉第反应的存在也更为明显(图9c)。在电化学剥离的石墨烯中,充-放电曲线显示出对称的三角形形状,而在g-nbd电极中,曲线偏离理想的线性形状,并且观察到大约0.2v和大约0.6v处的两个平稳区域,这两个平稳区域对应于氧化还原反应。
g-bbd主要显示法拉第反应,该法拉第反应在研究的电势范围内具有急剧的氧化电流和还原电流。这可能是由于溴基团的氧化还原反应。因此,对于超级电容器技术中的应用,与nbd相比,溴-官能化的石墨烯为次优选的。
从0.1vs-1的cv计算每个电极的比电容,并且对于基于电化学剥离的石墨烯电极获得19fg-1的值。与基于电化学剥离的石墨烯基电极相比,如本文所述的具有官能化石墨烯的电极显示出更大的比电容。电容随着官能化程度的增加而增加,这被认为是由于法拉第反应的贡献。在电极中分别获得了31.4fg-1、56.9fg-1和71.3fg-1的比电容值,该电极分别在1mm、40mm和100mmnbd中官能化。
尽管与基于电化学剥离的石墨烯电极相比,基于官能化石墨烯电极的总电容(双电层电容和伪电容)增加超过三倍,但是从法拉第反应的贡献中获得了显著比例的电容增量。对cv的仔细观察还表明,使用40mmg-nbd和1mmg-nbd电极测量的充电电流没有任何显著量的变化。这些观察结果表明石墨烯的官能化优选发生在边缘位置而不是基面,并且该论点得到了重新堆叠的官能化石墨烯的粉末x射线衍射数据的分析的支持。当获得粉末xrd数据时,g-nbd的(002)峰(26.6°处)实际上覆盖了电化学剥离的石墨烯的位置,这证明官能化没有改变层间距离。如果官能化发生在基面上,那么层间距离的变化是能够预期的。
使用aqd的官能化和剥离
还使用aqd执行了石墨的电化学剥离和官能化以生产官能化的石墨烯。aqd是指蒽醌-1-重氮。
石墨烯的电化学剥离和官能化使用三个电极装置来执行,该三个电极装置由等压成型的石墨棒/石墨箔工作电极、银线参比电极和等压成型的石墨棒对电极组成。暴露于电解质的工作电极的有效面积为大约12cm2。电解质通过在无水dmso中溶解0.3mcsclo4和15mm的蒽醌重氮(anthraqunonediazonium,aqd)来制备。同时的电化学剥离和官能化使用计时电流法、通过在恒定搅拌下施加相对于ag线的-4.0v的电势2小时来执行。然后剥离的产物用大量丙酮和超纯水来洗涤,并在60℃下在真空中干燥过夜。官能化粉末通过超声处理30分钟分散在期望的溶剂(nmp、水、异丙醇、以及水与异丙醇的混合物)中。得到的混合物在4000rpm下离心30分钟,上清液使用移液管在不扰乱滤渣的情况下提取。
剥离产物的表征
公布的电化学表征证明了少量层官能化石墨烯的形成(图10和11)。在图10中,获得了石墨的虚(低)线拉曼光谱,同时获得了40mm浓度的产物的实(高)线拉曼光谱。图11比较了石墨烯(内曲线)和生产的aqd-官能化石墨烯(外曲线)。外曲线中的峰的存在是aqd的典型特征。
aqd-cl的电化学
使用循环伏安法研究了adq的电化学行为(图12)。提出了以下反应顺序。c1归因于蒽醌自由基的形成,该蒽醌自由基与石墨表面迅速反应,c2归因于可逆质子转移到adq氧基团上。c3是由于cs+的嵌入,这表明在石墨表面上adq的存在不会阻碍cs+的插入。
图13显示了产物的示意性视图。与g-ngb和g-bbd不同,g-adq产物在水中不可溶。这归因于蒽醌结构中酮基团的疏水性。其在nmp中为可溶的。
***
本发明包括描述的多个方面和优选特征的组合,除非这样的组合是明显地不允许的或清楚地避免的。
为避免任何疑问,本文提供的任何理论解释提供为用于提高读者的理解的目的。发明人不希望受任何这些理论解释的束缚。
本文使用的章节标题仅是为了组织目的,且不应解释为限制所描述的主题。
在整个说明书中,包括随后的权利要求书,除非上下文另有要求,否则词语“包括(comprise)”和“包含(include)”,且诸如“包括(comprises)”、“包括(comprising)”和“包含(including)”的变体将被理解为暗示包括所述的整体或步骤、或整体或步骤的组,但不排除任何其他整体或步骤、或整体或步骤的组。
必须注意的是,除非上下文另有明确说明,否则如说明书和所附权利要求中所使用的,单数形式“一(a)”、“一个(an)”和“该(the)”包括复数指示物。范围可在本文表示为从“约”一个特定值,和/或到“约”另一个特定值。当表示这样的范围时,另外的实施方案包括从一个特定值和/或到另一个特定值。类似地,当值通过使用先行词“约”表示为近似值时,应当理解该特定值形成另一个实施方案。
参考文献
本文引用以下文献。各文献出于所有目的通过引用并入本文。
abdelkader,a.m.;kinloch,i.a.;dryfe,r.a.w.,continuouselectrochemicalexfoliationofmicrometer-sizedgrapheneusingsynergisticionintercalationsandorganicsolvents.acsappl.mater.interfaces2014,6,1632-1639.
allongue,p.;delamar,m.;desbat,b.;fagebaume,o.;hitmi,r.;pinson,j.;saveant,j.m.,covalentmodificationofcarbonsurfacesbyarylradicalsgeneratedfromtheelectrochemicalreductionofdiazoniumsalts.j.am.chem.soc.1997,119,201-207.
bissett,m.a.;kinloch,i.a.;dryfe,r.a.w.,characterizationofmos2-graphenecompositesforhigh-performancecoincellsupercapacitors.acsappl.mater.interfaces2015,7,17388-17398.
englert,j.m.;dotzer,c.;yang,g.a.;schmid,m.;papp,c.;gottfried,j.m.;steinruck,h.p.;spiecker,e.;hauke,f.;hirsch,a.,covalentbulkfunctionalisationofgraphene.
greenwood,j.;phan,t.h.;fujita,y.;li,z.;lvasenko,o.;vanderlinden,w.;vangorp,h.;frederickx,w.;lu,g.;tahara,k.;tobe,y.;uji-i,h.;mertens,s.f.l.;defeyter,s.,covalentmodificationofgrapheneandgraphiteusingdiazoniumchemistry:tunablegraftingandnanomanipulation.acsnano2015,9,5520-5535.
hernandez,y.;nicolosi,v.;lotya,m.;blighe,f.m.;sun,z.;de,s.;mcgovern,i.t.;holland,b.;byrne,m.;gun'ko,y.k.;boland,j.j.;niraj,p.;duesberg,g.;krishnamurthy,s.;goodhue,r.;hutchison,j.;scardaci,v.;ferrari,a.c.;coleman,j.n.,high-yieldproductionofgraphenebyliquid-phaseexfoliationofgraphite.nat.nanotechnol.2008,3,563-568.
kim,j.;kwon,s.;cho,d.-h.;kang,b.;kwon,h.;kim,y.;park,s.o.;jung,g.y.;shin,e.;kim,w.-g.;lee,h.;ryu,g.h.;choi,m.;kim,t.h.;oh,j.;park,s.;kwak,s.k.;yoon,s.w.;byun,d.;lee,z.;lee,c.,directexfoliationanddispersionoftwo-dimensionalmaterialsinpurewaterviatemperaturecontrol.nat.commun.2015,6.
konios,d.;stylianakis,m.m.;stratakis,e.;kymakis,e.,dispersionbehaviourofgrapheneoxideandreducedgrapheneoxide.j.colloidinterfacesci.2014,430,108-112.
lim,h.;lee,j.s.;shin,h.-j.;shin,h.s.;choi,h.c.,spatiallyresolvedspontaneousreactivityofdiazoniumsaltonedgeandbasalplaneofgraphenewithoutsurfactantanditsdopingeffect.langmuir2010,26,12278-12284.
lotya,m.;king,p.j.;khan,u.;de,s.;coleman,j.n.,high-concentration,surfactant-stabilizedgraphenedispersions.acsnano2010,4,3155-3162.
mendes,p.;belloni,m.;ashworth,m.;hardy,c.;nikitin,k.;fitzmaurice,d.;critchley,k.;evans,s.;preece,j.,anovelexampleofx-ray-radiation-inducedchemicalreductionofanaromaticnitro-group-containingthinfilmonsio2toanaromaticaminefilm.chemphyschem2003,4,884-889.
mu,y.;yu,h.q.;zheng,j.c.;zhang,s.j.;sheng,g.p.,reductivedegradationofnitrobenzeneinaqueoussolutionbyzero-valentiron.chemosphere2004,54,789-794.
niyogi,s.;bekyarova,e.;itkis,m.e.;zhang,h.;shepperd,k.;hicks,j.;sprinkle,m.;berger,c.;lau,c.n.;deheer,w.a.;conrad,e.h.;haddon,r.c.,spectroscopyofcovalentlyfunctionalisedgraphene.nanolett.2010,10,4061-4066.
saby,c.;ortiz,b.;champagne,g.y.;belanger,d.,electrochemicalmodificationofglassycarbonelectrodeusingaromaticdiazoniumsalts.1.blockingeffectof4-nitrophenyland4-carboxyphenylgroups.langmuir1997,13,6805-6813.
silvester,d.s.;wain,a.j.;aldous,l.;hardacre,c.;compton,r.g.,electrochemicalreductionofnitrobenzeneand4-nitrophenolintheroomtemperatureionicliquidc(4)dmimn(tf)(2).j.electroanal.chem.2006,596,131-140.
solis-fernandez,p.;bissett,m.a.;tsuji,m.;ago,h.,tunabledopingofgraphenenanoribbonarraysbychemicalfunctionalisation.nanoscale2015,7,3572-3580
smith,w.h.;bard,a.j.,electrochemicalreactionsoforganiccompoundsinliquidammonia.ii.nitrobenzeneandnitrosobenzene.j.am.chem.soc.1975,97,5203-5210.
stoller,m.d.;ruoff,r.s.,bestpracticemethodsfordetermininganelectrodematerial'sperformanceforultracapacitors.energyenviron.sci.2010,3,1294-1301.
sun,z.;kohama,s.-i.;zhang,z.;lomeda,j.r.;tour,j.m.,solublegraphenethroughedge-selectivefunctionalisation.nanoresearch2010,3,117-125.
zhong,y.l.;swager,t.m.,enhancedelectrochemicalexpansionofgraphiteforinsituelectrochemicalfunctionalisation.j.am.chem.soc.2012,134,17896-17899.
wo2012/120264
wo2013/132261
wo2015/019093
- 还没有人留言评论。精彩留言会获得点赞!