筛选靶向靶生物实体的候选生物实体的方法与流程
本申请要求于2013年6月13日提交的美国临时申请号61/834,782的权益,该临时申请通过引用全文并入本文。
发明背景
基因组学中的高通量技术和生物筛选、组合化学以及计算机技术已经彻底革新了发现药物以治疗疾病的工作。但是,科学家在寻找有效药物时仍必须在庞大数量的潜在候选药物中进行筛选。
使用传统筛选技术来鉴定别构调节剂是一项艰巨的任务。在所有蛋白质中只有约15%在活性位点具有足够深的口袋(pocket)来比天然配体更紧密地支持抑制剂的结合。对靶标如酶的活性测定可以并已经用于鉴定别构调节剂,但作用机制可能是难以确定的,因为这些测定通常涉及多种蛋白质,并且调节剂的作用可能针对除感兴趣的靶标之外的其他蛋白质。这些调节剂的筛选包括从非竞争性抑制剂中分选竞争性抑制剂,然后从后一组中的非特异性调节剂(聚集剂、高化学计量的结合剂等)中分选别构调节剂。
技术实现要素:
本发明提供了筛选靶向靶生物分子的候选药物的方法,其包括根据第一信号与第二信号之间的信号差异选择候选药物。该信号差异指示体内或体外药理学性质的差异。所述第一信号在通过使靶生物分子与第一候选物接触而使靶生物分子与第一候选物结合时产生,而所述第二信号在通过使靶生物分子与第二候选物接触而使靶生物分子与第二候选物结合时产生。采用表面选择性技术由二次谐波产生活性部分产生所述第一信号和所述第二信号。第一分子与第二分子具有相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,在选择步骤之前,所述方法还包括使靶生物分子与第一候选物接触,以在靶生物分子与第一候选物结合时检测第一信号;以及使靶生物分子与第二候选物接触,以在靶生物分子与第二候选物结合时检测第二信号。任选地,第一分子与第二分子具有相同的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,所述方法还包括产生第一信号与第二信号之间的信号差异。任选地,该信号差异为高于噪声水平1%或更多。任选地,该信号差异为高于噪声水平10%或更多。任选地,第二候选物是与期望的体内或体外药理学性质有关的参考分子。任选地,正信号差异表明体内或体外药理学性质的改善。任选地,负信号差异表明体内或体外药理学性质的改善。任选地,该信号是浓度归一化的。任选地,该信号可以是归一化的二次谐波产生强度。任选地,所述方法还包括在动物中筛选所选定的候选药物。
本发明还提供了筛选靶向靶生物分子的候选药物的方法,其包括根据信号选择候选药物,该信号是在靶生物分子与候选分子的结合复合物和与候选分子靶向相同结合位点的参考分子接触时产生的。该参考分子与体内或体外药理学性质有关。所述信号采用表面选择性技术由二次谐波产生活性部分产生。所述候选分子与所述参考分子具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,在选择步骤之前,所述方法还包括:使靶生物分子与候选分子接触,其中该候选分子结合至靶生物分子中的结合位点,由此形成结合复合物;使该结合复合物与参考分子接触;以及检测该结合复合物与参考分子接触时的信号。任选地,高于噪声水平不超过1%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,高于噪声水平不超过10%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,该信号是浓度归一化的。任选地,该信号可以是归一化的二次谐波产生强度。任选地,所述方法还包括在动物中筛选所选定的候选药物。
本发明还提供了筛选靶向靶生物分子的候选药物的方法,其包括根据信号选择候选药物,该信号是在靶生物分子与参考分子的结合复合物和与参考分子靶向相同结合位点的候选分子接触时产生的。该参考分子与体内或体外药理学性质有关。所述信号采用表面选择性技术由二次谐波产生活性部分产生。第一候选物与参考分子具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,在选择步骤之前,所述方法还包括使靶生物分子与参考分子接触,其中该参考分子结合至靶生物分子中的结合位点,由此形成结合复合物;使该结合复合物与候选分子接触;以及检测该结合复合物与候选分子接触时的信号。任选地,高于噪声水平超过1%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,高于噪声水平超过10%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,该信号是浓度归一化的。任选地,该信号可以是归一化的二次谐波产生强度。任选地,所述方法还包括在动物中筛选所选定的候选药物。
本发明还提供了用于鉴别针对靶生物分子的结合部分的方法,其包括通过比较第一候选物与第二候选物的结构鉴别该结合部分,所述候选物根据第一信号与第二信号之间的信号差异来选择。该信号差异表明第一分子中存在结合部分,并且第二分子中不存在结合部分。所述第一信号通过使靶生物分子与第一分子接触而使靶生物分子与第一分子结合时产生。所述第二信号通过使靶生物分子与第二分子接触而使靶生物分子与第二分子结合时产生。采用表面选择性技术由二次谐波产生活性部分产生所述第一信号和所述第二信号。第一分子与第二分子具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,该信号差异指示体内或体外药理学性质的差异。任选地,该信号差异为高于噪声水平1%或更多。任选地,该信号差异为高于噪声水平10%或更多。任选地,在鉴别步骤之前,所述方法还包括使靶生物分子与第一分子接触,以在靶生物分子与第一分子结合时检测第一信号;以及使靶生物分子与第二分子接触,以在靶生物分子与第二分子结合时检测第二信号。任选地,所述方法还包括产生第一信号与第二信号之间的信号差异。任选地,该信号是浓度归一化的。任选地,该信号可以是归一化的二次谐波产生强度。
本发明还提供了用于鉴别针对靶生物分子的结合部分的方法,其包括通过比较候选分子和参考分子的结构来鉴别该结合部分,该候选分子和参考分子根据靶生物分子与候选分子的结合复合物和与候选分子靶向相同结合位点的参考分子接触时产生的信号来选择。该信号表明参考分子中存在结合部分,并且候选分子中不存在结合部分。该信号采用表面选择性技术由二次谐波产生活性部分产生。所述候选分子与所述参考分子具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,高于噪声水平不超过1%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,高于噪声水平不超过10%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,在鉴别步骤之前,所述方法还包括使靶生物分子与候选分子接触,其中该候选分子结合至靶生物分子中的结合位点,由此形成结合复合物;使该结合复合物与参考分子接触;以及检测该结合复合物与参考分子接触时的信号。任选地,该信号是浓度归一化的。任选地,该信号可以是归一化的二次谐波产生强度。
本发明还提供了用于鉴别针对靶生物分子的结合部分的方法,其包括通过比较候选分子和参考分子的结构来鉴别该结合部分,该候选分子和参考分子根据靶生物分子与参考分子的结合复合物和与参考分子靶向相同结合位点的候选分子接触时产生的信号来选择。该信号表明参考分子中存在结合部分,并且候选分子中不存在结合部分。该信号采用表面选择性技术由二次谐波产生活性部分产生。所述候选分子与所述参考分子具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。任选地,高于噪声水平超过1%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,高于噪声水平超过10%的信号指示与参考分子相比相当的或增加的体内或体外药理学性质。任选地,在鉴别步骤之前,所述方法还包括使靶生物分子与参考分子接触,其中该参考分子结合至靶生物分子中的结合位点,由此形成结合复合物;使该结合复合物与候选分子接触;以及检测该结合复合物与候选分子接触时的信号。任选地,该信号是浓度归一化的。任选地,该信号可以是归一化的二次谐波产生强度。
任选地,所述体内药理学性质是治疗效力。任选地,该体内药理学性质是临床效力性质。任选地,该体内药理学性质是药代动力学性质。任选地,该体内药理学性质是临床毒性性质。任选地,该体内药理学性质是临床前毒性性质。任选地,该体内药理学性质是药效学性质。任选地,该体内药理学变量是治疗指数。任选地,所述结合位点是活性位点。任选地,该结合位点是别构结合位点。任选地,所述靶生物分子是蛋白质。任选地,该靶生物分子是DNA。任选地,该靶生物分子是α突触核蛋白。任选地,所述方法还包括在动物中筛选所选定的候选药物。任选地,该候选药物是小分子。任选地,该候选药物是蛋白质。任选地,该候选药物是抗体。任选地,该候选药物是生物改良药或生物仿制药。任选地,该候选药物是四磺酸酞菁衍生物或类似物。任选地,该候选药物是针对α突触核蛋白的抗体。任选地,该候选药物是苏兰珠单抗(solanezumab)或巴匹珠单抗(bapineuzumab)的生物等价物。
本公开内容提供了筛选生化实体的方法,该方法包括:(a)提供包含6种或更多种候选生化实体的文库,其中如采用一个或多个结构相似性参数确定的,该6种或更多种候选生化实体是结构上相似的,该结构相似性参数选自:(i)各种候选生化实体的分子量彼此在150g/mol以内;(ii)各种候选生化实体的分子量彼此在30%以内;(iii)各种候选生化实体的化学式彼此在15个原子以内;(iv)各种候选生化实体之间的原子总数之差小于15;(v)碳原子数之差小于5;(vi)氢原子数之差小于5;(vii)氮原子数之差小于5;(viii)氧原子数之差小于5;(ix)硫原子数之差小于5;(x)如通过log D测量的,各种候选生物实体的极性彼此在10%以内;(xi)如通过log P(分配系数)测量的,各种候选生物实体的极性彼此在10%以内;(xii)如通过PSA(极性表面积)测量的,各种候选生物实体的极性彼此在10%以内;(xiii)相同的骨架核心;(xiv)如通过图论测量的化学相似性;(xv)Tanimoto(或Jaccard)系数T大于0.85;(xvi)芳香族基团数目之差小于3;(xvii)杂环基团数目之差小于3;(xviii)单环基团数目之差小于3;(xix)稠环体系数目之差小于3;以及(xx)双键数目之差小于3;(b)测量靶生化实体的第一构象状态,由此产生基线;(c)使各种候选生化实体与相同的靶生化实体接触;(d)测量靶生化实体的第二构象状态;(e)采用第二构象状态确定靶生化实体的信号特征(signature);以及(f)基于该信号特征从候选生化实体中选择一种或多种生化实体。
在一些情况下,可采用全内反射(TIR)测量第一或第二构象状态。在一些情况下,可采用二次谐波产生(SHG)测量第一或第二构象状态。
在一些情况下,确定信号特征可包括测量靶生化实体的构象状态的动力学(实时)变化。在其他情况下,确定信号特征可包括测量靶生化实体的构象状态的终点变化。在又一些其他情况下,确定信号特征可包括测量信号的信号强度。在进一步的情况下,信号特征的确定使用基线。
在一些情况下,所述方法还可包括根据一个或多个结构相似性参数,采用计算机可执行程序分析所述6种或更多种候选生化实体。在其他情况下,所述方法还可包括采用计算机可执行程序分析所述信号特征。
各种候选生化实体可产生靶生化实体的构象状态的不同变化。此外,当在步骤(c)中接触时,各种候选生化实体可以产生不同的信号特征。
所述方法还可包括将靶生化实体的构象状态的变化与由已知构象调节剂产生的构象状态变化进行比较。所述已知构象调节剂可以是已知的药物。所述方法还可包括基于与由已知构象调节剂诱导的构象状态变化最相似的构象状态变化来选择一种或多种生化实体。在一些情况下,由已知构象调节剂产生的构象状态变化可以与药理学性质或功能效果相关联。
在一些情况下,候选生化实体中的至少两种具有相似的药理学性质或功能效果。在其他情况下,候选生化实体中的至少两种不具有相似的药理学性质或功能效果。而且,可以根据信号特征与已知药理学性质或已知功能效果的相关性来选择一种或多种生化实体。
第一构象状态可在靶生化实体与物剂温育后测量。例如,该物剂可以是来自文库的候选生化实体。
所述方法还可包括测量选自下组的信号特征参数:i)信号特征的大小相对于基线的变化;ii)信号特征的方向相对于基线的变化;以及iii)候选生化实体接触时信号特征的单位时间信号率的变化。此外,如采用一个或多个信号特征相似性参数确定的,由第一候选生化实体产生的信号特征和由第二候选生化实体产生的信号特征可以是相似的,该信号特征相似性参数选自:i)接触第一候选生化实体时信号特征的大小相对于基线的变化与接触第二候选生化实体时信号特征的大小相对于基线的变化相差50%以内;ii)接触第一候选生化实体时信号特征的方向相对于基线的变化与接触第二候选生化实体时信号特征的方向相对于基线的变化相同;以及iii)接触第一候选生化实体时信号特征的单位时间信号率的变化与接触第二候选生化实体时信号特征的单位时间信号率的变化相差两倍以内。
本公开内容还提供了筛选生化实体的方法,该方法包括:(a)提供包含6种或更多种候选生化实体的文库;(b)测量靶生化实体的第一构象状态,由此产生基线;(c)使各种候选生化实体与相同的靶生化实体接触;(d)测量靶生化实体的第二构象状态;(e)采用第二构象状态确定至少第一和第二候选生化实体的信号特征;(f)将第一生化实体的信号特征与第二生化实体的信号特征进行比较;以及(g)根据(f)中的比较从候选生化实体中选择一种或多种生化实体。
在一些情况下,如采用一个或多个结构相似性参数确定的,所述6种或更多种候选生化实体是结构上相似的,该结构相似性参数选自:(i)各种候选生化实体的分子量彼此在150g/mol以内;(ii)各种候选生化实体的分子量彼此在30%以内;(iii)各种候选生化实体的化学式彼此在15个原子以内;(iv)各种候选生化实体之间的原子总数之差小于15;(v)碳原子数之差小于5;(vi)氢原子数之差小于5;(vii)氮原子数之差小于5;(viii)氧原子数之差小于5;(ix)硫原子数之差小于5;(x)如通过log D测量的,各种候选生物实体的极性彼此在10%以内;(xi)如通过log P(分配系数)测量的,各种候选生物实体的极性彼此在10%以内;(xii)如通过PSA(极性表面积)测量的,各种候选生物实体的极性彼此在10%以内;(xiii)相同的骨架核心;(xiv)如通过图论测量的化学相似性;(xv)Tanimoto(或Jaccard)系数T大于0.85;(xvi)芳香族基团数目之差小于3;(xvii)杂环基团数目之差小于3;(xviii)单环基团数目之差小于3;(xix)稠环体系数目之差小于3;以及(xx)双键数目之差小于3。
所述方法还可包括采用计算机可执行程序分析信号特征。各种候选生化实体可产生靶生化实体的不同的构象状态变化。在一些情况下,当在(c)中接触时,各种候选生化实体可以产生不同的信号特征。在一些情况下,一种或多种生化实体的信号特征与已知的药理学性质或已知的功能效果相关联。在进一步的情况下,至少两种候选生化实体具有相似的药理学性质或功能效果。
所述方法还可包括测量选自下组的参数:i)信号特征的大小相对于基线的变化;ii)信号特征的方向相对于基线的变化;以及iii)候选生化实体接触时信号特征的单位时间信号率的变化。此外,如采用一个或多个信号特征相似性参数确定的,由第一候选生化实体产生的信号特征和由第二候选生化实体产生的信号特征可以是相似的,该信号特征相似性参数选自:i)接触第一候选生化实体时信号特征的大小相对于基线的变化与接触第二候选生化实体时信号特征的大小相对于基线的变化相差50%以内;ii)接触第一候选生化实体时信号特征的方向相对于基线的变化与接触第二候选生化实体时信号特征的方向相对于基线的变化相同;以及iii)接触第一候选生化实体时信号特征的单位时间信号率的变化与接触第二候选生化实体时信号特征的单位时间信号率的变化相差两倍以内。
本公开内容还提供了生化实体编目方法,该方法包括:(a)提供生化实体的文库,其中各种生化实体包含不同的分子结构;(b)使该文库中的至少一种生化实体与相同的靶生化实体接触;(c)测量靶生化实体的构象状态;(d)采用该构象状态确定信号特征;以及(e)采用该信号特征产生结构-活性关系(SAR)目录。
在一些情况下,产生SAR可包括将信号特征与结合性质、功能性质、效力、效能、选择性或其组合相关联。在一些情况下,产生SAR可包括将信号特征与候选生化实体的一个或多个结构参数相关联,其中所述结构参数可选自:分子量、化学式、总原子数、碳原子数、氢原子数、氮原子数、氧原子数、硫原子数、如通过log D测量的极性、如通过log P测量的极性、如通过PSA(极性表面积)测量的极性、骨架核心、如通过图论测量的化学相似性、Tanimoto(或Jaccard)系数T、芳香族基团的数目、杂环基团的数目、单环基团的数目、稠环体系的数目以及双键的数目。
所述方法还可包括在接触之前测量靶生化实体的构象状态,由此产生基线。该方法还可包括分析SAR以确定至少一种生化实体内的结合部分,该结合部分与药理学性质或功能效果相关。
在一些情况下,确定信号特征可包括测量靶生化实体的构象状态的动力学(实时)变化。在其他情况下,确定信号特征包括测量靶生化实体的构象状态的终点变化。该信号特征可指示靶生化实体的构象状态的变化。所述方法还可包括采用计算机可执行程序分析信号特征。
生化实体的文库可包含候选药物。靶生化实体可以是药物靶标。该药物靶标可具有已知的药理学性质或已知的功能效果。所述方法还可包括根据SAR目录或信号特征与已知药理学性质或已知功能效果的相关性来选择一种或多种生化实体。
所述方法还可包括测量选自下组的信号特征参数:i)信号特征的大小相对于基线的变化;ii)信号特征的方向相对于基线的变化;以及iii)候选生化实体接触时信号特征的单位时间信号率的变化。此外,如采用一个或多个信号特征相似性参数确定的,由第一候选生化实体产生的信号特征和由第二候选生化实体产生的信号特征可以是相似的,该信号特征相似性参数选自:i)接触第一候选生化实体时信号特征的大小相对于基线的变化与接触第二候选生化实体时信号特征的大小相对于基线的变化的相差50%以内;ii)接触第一候选生化实体时信号特征的方向相对于基线的变化与接触第二候选生化实体时信号特征的方向相对于基线的变化相同;以及iii)接触第一候选生化实体时信号特征的单位时间信号率的变化与接触第二候选生化实体时信号特征的单位时间信号率的变化相差两倍以内。
援引并入
本说明书中提到的所有出版物、专利和专利申请均通过引用并入本文,其程度如同特别地且单独地指出每个单独的出版物、专利或专利申请均通过引用而并入。
附图简述
本发明的新颖特征在所附的权利要求书中具体阐述。通过参考以下对利用了本发明原理的说明性实施方案加以阐述的发明详述以及附图,将会获得对本发明的特征和优点的更好的理解,附图中:
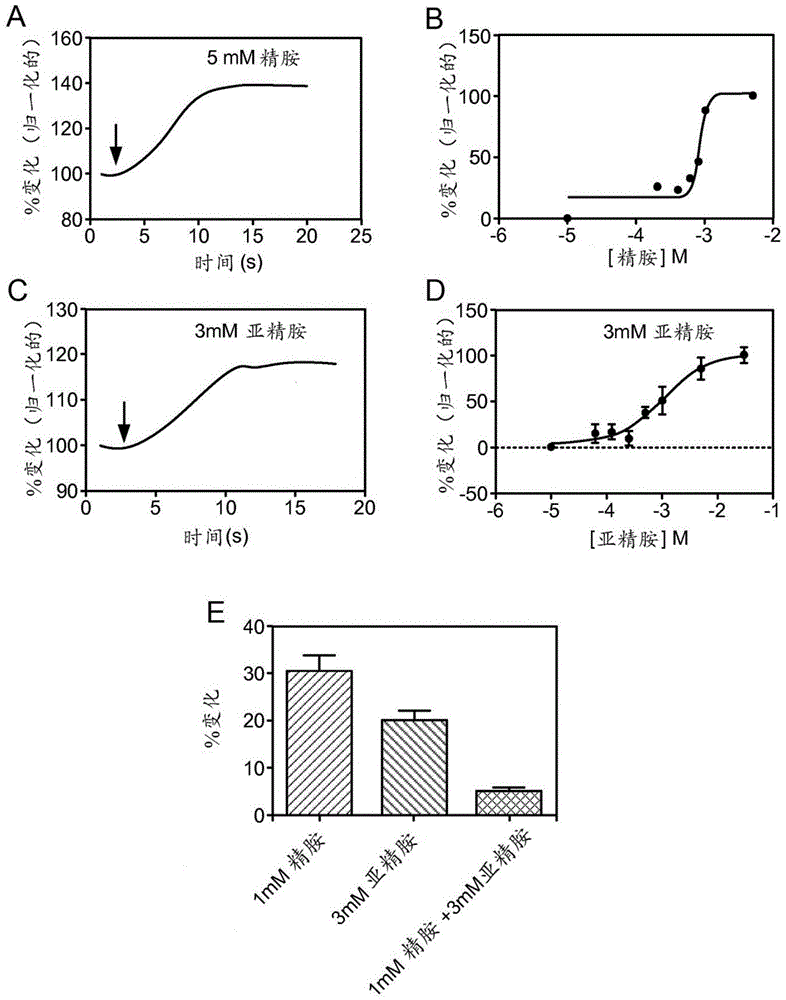
图1A示出了如通过二次谐波产生所测量的,精胺和亚精胺诱导α-突触核蛋白的构象变化。代表性迹线(trace)示出了α-突触核蛋白暴露于5mM精胺时的实时SHG响应。箭头表示精胺添加。SHG强度的变化相对于临注射前的值进行归一化。
图1B示出了如通过SHG测量的,精胺对α-突触核蛋白构象的剂量响应曲线。通过向5μMα-突触核蛋白添加给定浓度的精胺并测量SHG强度的变化来确定剂量曲线(以对数尺度作图)。每个浓度下SHG强度的变化被定量为变化(本文中也称为“改变”)百分比。
图1C提供了示出α-突触核蛋白暴露于3mM亚精胺时的实时SHG响应的代表性迹线。箭头表示亚精胺添加。SHG强度的变化相对于临注射前的值进行归一化。
图1D示出了如通过SHG测量的,亚精胺对α-突触核蛋白构象的剂量响应曲线。通过向5μMα-突触核蛋白添加给定浓度的亚精胺并测量SHG强度的变化来确定剂量曲线(以对数尺度作图)。每个浓度下SHG强度的变化被定量为变化百分比。误差条=SEM。N=3。
图1E是精胺-亚精胺竞争测定的定量。1mM精胺和3mM亚精胺对照分别示于左侧柱和中间柱。向与1mM精胺一起预温育的样品添加3mM亚精胺示于右侧柱。SHG强度的变化以变化百分比绘出。误差条=SEM。N=4。
图2A示出了Maybridge Ro3片段文库的基于二次谐波产生的筛选。代表性迹线示出了用于针对α-突触核蛋白的精胺诱导的构象变化的抑制剂筛查Maybridge Ro3片段文库的SHG响应。左侧为α-突触核蛋白暴露于5mM精胺的典型响应。右侧为与1mM化合物A一起预温育的α-突触核蛋白暴露于5mM精胺的响应,其指示出相对于基线可忽略的变化。
图2B示意性地图示了精胺(顶部)和化合物A(底部)的化学结构。
图2C提供了代表性迹线,其示出了用于针对α-突触核蛋白的精胺诱导的构象变化的抑制剂筛查Maybridge Ro3片段文库的SHG响应。左侧为示出了α-突触核蛋白暴露于5mM精胺的典型SHG响应的代表性迹线。中间为与1mM化合物A一起预温育的α-突触核蛋白在暴露于5mM精胺后的SHG响应,其指示出相对于基线可忽略的变化。右侧为示出了α-突触核蛋白暴露于1mM化合物A时的实时SHG响应的代表性迹线。箭头表示化合物A添加。SHG强度的变化相对于临注射前的值进行归一化。
图3A示出了化合物A的表征。代表性迹线示出了α-突触核蛋白暴露于1mM化合物A时的实时SHG响应。箭头表示化合物A添加。SHG强度的变化相对于临注射前的值进行归一化。
图3B提供了如通过SHG测量的,化合物A对α-突触核蛋白构象的剂量响应曲线。通过向5μMα-突触核蛋白添加给定浓度的化合物A并测量SHG强度的变化来确定剂量曲线(以对数尺度作图)。每个浓度下SHG强度的变化被定量为变化百分比。误差条=SEM。N=3。
图3C是精胺-化合物A竞争测定的定量。5mM精胺对照示于左侧。向与精胺一起预温育的样品添加1mM或100μM化合物A分别示于中间柱和右侧柱。SHG强度的变化以变化(本文中也称为“改变”)百分比绘出。
图3D是精胺-化合物A竞争测定的定量。仅5mM精胺和1mM化合物A的典型SHG强度变化百分比分别示于第一柱和第二柱。向与精胺一起预温育的样品添加1mM或100μM化合物A时的SHG强度变化分别示于第三柱和第四柱。与1mM化合物A一起温育后添加5mM精胺时的SHG强度变化示于第五柱。SHG强度的变化以变化百分比绘出。误差条=SEM。N=3。
图4A示出了化合物A是α-突触核蛋白的精胺诱导的构象变化的高度特异性抑制剂。如通过程序SimFinder预测的,来自Maybridge Ro3文库的得分前五的化合物A类似物的化学结构。
图4B是α-突触核蛋白暴露于得分前五的化合物A的化学类似物时的构象变化的定量。将每种类似物添加至含有5μMα-突触核蛋白的孔中,并测量SHG强度的变化。指示1mM精胺添加的典型SHG响应的代表性数据集被包括在内以用于比较的目的。SHG强度的变化以变化百分比绘出。误差条=SEM。N=3。
图4C是类似物-精胺竞争测定中α-突触核蛋白的构象变化的定量。将指定的化合物A类似物与5μMα-突触核蛋白一起预温育25分钟,并且测量暴露于1mM精胺时的SHG强度变化。指示化合物A+1mM精胺竞争测定的典型SHG响应的代表性数据集被包括在内以用于比较的目的。SHG强度的变化以变化百分比绘出。
图4D是α-突触核蛋白暴露于得分前五的化合物A的化学类似物时的构象变化的定量。将每种类似物以1mM的最终浓度添加至含有5μMα-突触核蛋白的孔中,并测量SHG强度的变化。指示1mM化合物A添加的典型SHG响应的代表性数据集被包括在内以用于比较的目的。SHG强度的变化以变化百分比绘出。误差条=SEM。N=3。
图4E提供了在类似物1(左侧)、类似物2(中间)和类似物5(右侧)以1:12摩尔比存在下,α-突触核蛋白的1H-15N共振的化学位移扰动分析。误差条=SEM。N=3。
图5A示出了四磺酸酞菁(一种α-突触核蛋白聚集的抑制剂)产生不同的SHG信号特征。代表性迹线示出了α-突触核蛋白暴露于10μM PcT时的实时SHG响应。箭头指示PcT添加的时间。SHG强度的变化以变化百分比绘出。误差=SEM。N=5。
图5B是α-突触核蛋白暴露于精胺、亚精胺和四磺酸酞菁时的SHG强度变化的定量。5mM精胺示于左侧,3mM亚精胺示于中间,而10μM PcT示于右侧。SHG强度的变化以变化百分比绘出。误差条=SEM。N=4。
图6A示出了2-D HSQC NMR表明化合物A与精胺结合至蛋白质的C末端区域上的相同位点。
图6B是在不存在(黑色)和存在(红色)化合物A的情况下,100μMα-突触核蛋白的1H-15N NMR谱的叠加详图。
图6C是α-突触核蛋白在以1:10摩尔比存在化合物A下的1H-15N共振的化学位移扰动分析。
图6D是在不存在(黑色)和存在(红色)化合物A(3mM)的情况下,MTSL-标记的A90Cα-突触核蛋白(100μM)中酰胺质子的顺磁弛豫增强图谱。
图7A示出了硫黄素T(Thioflavin T)试验表明化合物A使体外α-突触核蛋白的聚集速率增加大约两倍。
图7B是在存在(左侧)和不存在(右侧)化合物A的情况下获得的α-突触核蛋白原纤维的电子显微照片。
图8示出了添加多巴胺时α-突触核蛋白的SHG强度的动力学(实时)变化。
图9是仅添加缓冲液或添加1mM多巴胺时α-突触核蛋白的SHG强度的平均变化的定量。
具体实施方式
虽然本文已经示出并描述了本发明的多个实施方案,但对于本领域技术人员而言很明显,这些实施方案仅以示例的方式提供。在不背离本发明的情况下,本领域技术人员可想到许多变化、改变和替代。应该理解,可以采用本文所述的本发明实施方案的各种替代方案。应当理解,本发明的不同方面可以单独地、共同地或彼此组合地理解。
定义
如本文使用的“二次谐波”是指两倍于基本光束的频率的光频率。
如本文所使用的,“表面选择性”指非线性光学技术,例如二次谐波产生或者和频/差频产生,或者本领域已知的其他表面特异性技术。
如本文所使用的,“和频产生”(SFG)是一种非线性光学技术,通过该技术,一个频率(Ω1)的光与另一频率(Ω2)的光混合,以产生在和频(Ω1+Ω2)处的响应(Yang,G,Shen,YR,Optics Letters,9(11):510-512,1984;Shen,YR,Annual Review of Physical Chemistry 40(1):327-350,1989)。例如,SFG尤其可用于通过分子的特征性振动跃迁来检测表面处的分子,在这种情况下,SFG本质上是一种具有在可见光和红外线频率的Ω1和Ω2的表面选择性红外光谱法。当术语“SHG”或“二次谐波产生”在本文中使用时,应当理解,“SFG”与“和频产生”可采用本领域技术人员公知的方法替换SHG,并代替SHG使用。
如本文所使用的“非线性活性部分”是具有超极化性的物质。
如本文所使用的“二次谐波活性部分”是指非线性活性部分、粒子或分子,其可连接或并入(例如,共价或非共价地连接,通常是并入,等等)至分子(例如,蛋白质,如酶)、粒子、细胞或相(例如,脂双层),从而使其具有(例如,更高的)非线性光学活性。
如本文所使用的“别构”、“别构调节剂”或“别构候选物”是指这样的分子、部分或物质:其主要与活性位点以外的位点结合并可引起如通过SHG或SFG确定的构象变化(例如,在一个或多个邻近结合位点的位置和/或在一个或多个不邻近结合位点的位置),从而通过别构作用机制发挥其作用。
如本文所使用的“活性位点”或“活性结合位点”是指这样的靶蛋白区域,由于其形状和电荷电势,该区域有利地通过各种共价和/或非共价结合力与另一物剂(例如,配体、蛋白质、多肽、肽、核酸(包括DNA或RNA)、分子、化合物、抗生素或药物、分子、部分、底物、产物、类似物或抑制剂)相互作用或相关联,并且在其中行使该蛋白质的功能(例如催化、信号传导和/或效应物活化)。
如本文所定义的,术语“配体”包括与另一分子结合的任何分子,例如,与另一蛋白质结合的一种蛋白质、与蛋白质结合的碳水化合物或者与蛋白质结合的小分子。
如本文所使用的,“非线性”是指能够转变入射光束(也称为基本光束)的频率的光学技术。非线性光束是由这样的转变所产生的光束(例如,更高阶频率的光束),例如二次谐波。在二次谐波、和频或差频产生中,非线性光束相干地(coherently)产生。在二次谐波产生(SHG)中,基本光束(fundamental beam)的两个光子被界面虚能地(virtually)散射,从而产生二次谐波的一个光子。在本文中也称为“非线性光学的”或“表面选择性非线性的”。
如本文所使用的术语“非线性活性的”或“非线性地活性的”也指分子、粒子、界面或相在受到一个或多个入射辐射束驱动时产生非线性光辐射的能力的一般性质。
当本文中提及非线性光学方法时,“检测”是指这样的技术,通过该技术,表面选择性非线性光辐射的性质可用于检测、测量探针-靶标相互作用(例如蛋白质与候选化合物之间的相互作用)的性质或该相互作用的效应,或将该性质或效应与非线性光学光的性质(例如,强度、波长、偏振或其他与电磁辐射所共有的性质)相关联。
目的在于在整个说明书中给出的每一最大数值限值包括每个较低的数值限值,如同这样的较低的数值限值在此明确地写出。在整个说明书中给出的每一最小数值限值包括每个较高的数值限值,如同这样的较高的数值限值在此明确地写出。整个说明书中给出的每一数值范围将包括每一个落在该较宽数值范围内的较窄数值范围,如同这样的较窄数值范围均在此明确地写出。
介绍
本文提供了用于筛选靶向靶生化实体的候选生化实体和/或结合部分(例如,与候选生化实体相关的一个或多个结合部分),例如,与体内或体外药理学性质相关的候选生化实体和/或结合部分的方法。这些方法均基于采用表面选择性技术由二次谐波产生(SHG)活性部分、和频产生(SFG)活性部分或差频产生(DFG)活性部分(例如,附接至候选物、靶标或两者)产生的信号。该方法可包括将两种或更多种候选物在靶标与不同候选物结合时产生的信号进行比较。在一些情况下,该候选物可以是具有相同或相似骨架核心的生化实体(例如,分子)。在一些情况下,该生化实体可包括靶向靶生物分子的候选药物(和/或与之相关的一个或多个结合部分)。
在一些实施方案中,本文的方法可用于筛选靶向靶生物分子的候选药物(和/或与之相关的结合部分),例如,与体内或体外药理学性质相关的候选物或结合部分。在一些情况下,该候选药物可以是具有相同或相似骨架核心的分子。本公开内容中与候选药物(和/或与之相关的结合部分)及靶生物分子相关地描述的任何方面均可至少在一些配置下同等地应用于任何类型的候选生化实体(和/或结合部分)以及任何类型的靶生化实体。例如,本公开内容可应用于细胞。而且,本公开内容中与参考分子相关地描述的任何方面均可至少在一些配置下同等地应用于任何类型的参考生化实体。
普遍认为,结构相似性导致某种程度的功能相似性。然而,单独的结构相似性并不总是与某些功能,特别是体内药理学性质相关联。因此,必须采用多种体内或体外试验对通过基于结构的设计而产生的候选药物进行广泛筛选以鉴别先导化合物。这一过程是冗长、费时、昂贵的且通常是徒劳的,并且已成为现代药物发现中的主要瓶颈之一。
在一个方面,本公开内容提供了筛选生化实体的方法,该方法包括:(a)提供包含多种候选生化实体的文库,其中如采用一个或多个结构相似性参数确定的,所述候选生化实体是结构上相似的;(b)测量靶生化实体的第一构象状态,由此产生基线;(c)使各种候选生化实体与靶生化实体接触;(d)测量靶生化实体的第二构象状态;(e)采用第二构象状态确定靶生化实体的信号特征;以及(f)基于该信号特征从候选生化实体中选择一种或多种生化实体。
在另一个方面,本公开内容提供了筛选生化实体的方法,该方法包括:(a)提供包含多种候选生化实体的文库;(b)测量靶生化实体的第一构象状态,由此产生基线;(c)使各种候选生化实体与靶生化实体接触;(d)测量靶生化实体的第二构象状态;(e)采用第二构象状态确定至少第一和第二候选生化实体的信号特征;(f)将第一生化实体的信号特征与第二生化实体的信号特征进行比较;以及(g)根据(f)中的比较从候选生化实体中选择一种或多种生化实体。
在又一方面,本公开内容提供了生化实体编目方法,该方法包括:(a)提供生化实体的文库,其中各种生化实体包含不同的分子结构;(b)使该文库中的至少一种生化实体与相同的靶生化实体接触;(c)测量靶生化实体的构象状态;(d)采用该构象状态确定信号特征;以及(e)采用该信号特征产生结构-活性关系(SAR)目录。
在一些情况下,产生SAR可包括将信号特征与结合性质、功能性质、效力、效能、选择性或其组合相关联。在一些情况下,产生SAR可包括将信号特征与候选生化实体的一个或多个结构参数相关联,其中所述结构参数选自:分子量、化学式、总原子数、碳原子数、氢原子数、氮原子数、氧原子数、硫原子数、如通过log D测量的极性、如通过log P测量的极性、如通过PSA(极性表面积)测量的极性、骨架核心、如通过图论测量的化学相似性、Tanimoto(或Jaccard)系数T、芳香族基团的数目、杂环基团的数目、单环基团的数目、稠环体系的数目以及双键的数目。
所述方法还可包括分析SAR以确定至少一种生化实体内与药理学性质或功能效果相关的结合部分。在一些情况下,可在接触之前测量靶生化实体的构象状态,从而产生基线。
所述文库可包含2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、22、24、26、28、30、35、40、45、50、60、70、80、90、100、120、140、160、180、200、250、300、350、400、450、500、600、700、800、900、1000、1200、1400、1600、1800、2000、2500、3000、3500、4000、4500、5000、6000、7000、8000、9000或10000种或者更多种候选生化实体。在一些实例中,该文库可包含6种或更多种候选生化实体。在进一步的实例中,该文库可包含20种或更多种候选生化实体。另外,该生化实体文库可包含候选药物。
如采用1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20个或更多个结构相似性参数所确定的,候选生化实体可以是结构上相似的。在一些实例中,如采用3个或更多个结构相似性参数所确定的,候选生化实体可以是结构上相似的。在进一步的实例中,如采用5个或更多个结构相似性参数所确定的,候选生化实体可以是结构上相似的。结构相似性参数的实例包括但不限于:
(i)各种候选生化实体的分子量彼此在10、20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、290、300g/mol以内;
(ii)各种候选生化实体的分子量彼此在1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、22%24%、26%、28%、30%、35%、40%、45%或50%以内;
(iii)各种候选生化实体的化学式彼此在1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、22、24、26、28、30、35、40、45、50、60、70、80、90、100、120、140、160、180、200、250、300、350、400、450、500、600、700、800、900或1000个原子以内;
(iv)各种候选生化实体之间的原子总数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、22、24、26、28、30、35、40、45、50、60、70、80、90、100、120、140、160、180、200、250、300、350、400、450、500、600、700、800、900或1000;
(v)碳原子数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
(vi)氢原子数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
(vii)氮原子数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
(viii)氧原子数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
(ix)硫原子数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
(x)如通过log D测量的,各种候选生化实体的极性彼此在1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、22%、24%、26%、28%、30%、35%、40%、45%或50%以内;
(xi)如通过log P(分配系数)测量的,各种候选生化实体的极性彼此在1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、22%、24%、26%、28%、30%、35%、40%、45%或50%以内;
(xii)如通过PSA(极性表面积)测量的,各种候选生化实体的极性彼此在1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、22%、24%、26%、28%、30%、35%、40%、45%或50%以内;
(xiii)相同的骨架核心;
(xiv)如通过图论测量的化学相似性;
(xv)Tanimoto(或Jaccard)系数T大于0.5、0.6、0.7、0.75、0.8、0.81、0.82、0.83、0.84、0.85、0.86、0.87、0.88、0.89、0.9、0.91、0.92、0.93、0.94、0.95、0.96、0.97、0.98或0.99;
(xvi)芳香族基团的数目之差小于1、2、3、4、5、6、7、8、9或10;
(xvii)杂环基团的数目之差小于1、2、3、4、5、6、7、8、9或10;
(xviii)单环基团的数目之差小于1、2、3、4、5、6、7、8、9或10;
(xix)稠环体系的数目之差小于1、2、3、4、5、6、7、8、9或10;
以及
(xx)双键的数目之差小于1、2、3、4、5、6、7、8、9或10。
在一些情况下,靶生化实体可以是固有无序蛋白质。例如,该固有无序蛋白质可以是α突触核蛋白。在一些情况下,靶生化实体可以是药物靶标。在一些情况下,靶生化实体可以不具有已知的构象调节剂。
在一些情况下,第一构象状态可在靶生化实体与物剂温育后测量。例如,该物剂可以是来自该文库的候选生化实体。
在一些情况下,构象状态可采用全内反射(TIR)测量。在一些情况下,构象状态可采用二次谐波产生(SHG)测量。
在一些情况下,确定信号特征可包括测量靶生化实体的构象状态的动力学(实时)变化。在其他情况下,确定信号特征可包括测量靶生化实体的构象状态的终点变化。在其他情况下,确定信号特征可包括测量信号特征的信号强度。在进一步的情况下,信号特征的确定可使用基线。此外,信号特征可指示靶生化实体的构象状态的变化。
在一些情况下,可采用计算机可执行程序根据一个或多个结构相似性参数来分析候选生化实体。在其他情况下,可采用计算机可执行程序分析信号特征。
各种候选生化实体可产生靶生化实体的构象状态的不同变化。而且,各种候选生化实体可产生不同的信号特征。
在一些情况下,还可将靶生化实体的构象状态的变化与由已知构象调节剂产生的构象状态的变化进行比较。在一些情况下,已知构象调节剂可以是已知的药物。在一些情况下,可根据与由已知构象调节剂诱导的构象状态变化最相似的构象状态变化来选择生化实体。此外,由已知构象调节剂产生的构象状态变化可以与药理学性质或功能效果相关联。
在一些情况下,一种或多种生化实体的信号特征或SAR目录可以与已知的药理学性质或已知的功能效果相关联。还可基于信号特征或SAR目录与已知药理学性质或已知功能效果的相关性来选择一种或多种生化实体。
在一些情况下,至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、22、24、26、28、30、35、40、45、50、60、70、80、90或100种候选生化实体具有相似的药理学性质或功能效果。在其他情况下,至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、22、24、26、28、30、35、40、45、50、60、70、80、90或100种候选生化实体不具有相似药理学性质或功能效果。
在本公开内容中提供的方法还可包括测量信号特征参数。信号特征参数的实例包括但不限于:
i)信号特征的大小相对于基线的变化;
ii)信号特征的方向相对于基线的变化;以及
iii)接触候选生化实体时信号特征的单位时间信号率的变化。
在一些情况下,如采用1、2、3个或更多个信号特征相似性参数确定的,由第一候选生化实体产生的信号特征与由第二候选生化实体产生的信号特征可以是相似的。信号特征相似性参数的实例包括但不限于:
i)接触第一候选生化实体时信号特征的大小相对于基线的变化与接触第二候选生化实体时信号特征的大小相对于基线的变化相差1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、22%24%、26%、28%、30%、35%、40%、45%或50%以内;
ii)接触第一候选生化实体时信号特征的方向相对于基线的变化与接触第二候选生化实体时信号特征的方向相对于基线的变化相同;以及
iii)接触第一候选生化实体时信号特征的单位时间信号率的变化与接触第二候选生化实体时信号特征的单位时间信号率的变化相差1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%、15%、16%、17%、18%、19%、20%、22%24%、26%、28%、30%、35%、40%、45%、50%、60%、70%、80%、90%、100%(一倍)、110%、120%、130%、140%、150%、160%、170%、180%、190%或200%(两倍)以内。
如在以下实例中详述的,本发明提供了将由候选物产生的SHG信号(或SFG信号、DFG信号)与其体内或体外药理学性质(特别是其体内性质)相关联的方法。因此,本发明提供了有效筛选具有一种或多种期望的体内或体外药理学性质的候选药物的方法,从而避免了对每种候选物进行昂贵的体内或体外测定的需要。
I.筛选靶向靶分子的候选药物的方法
本文提供了用于筛选靶向靶生物分子的候选药物的方法。该方法可用于筛选任何候选药物,例如,具有相同或相似的靶向靶生物分子中的结合位点的骨架核心的候选药物。
A.具有相同或相似骨架的候选药物
在小分子药物的情况下,“骨架”可以指小的靶标结合分子,一个或多个其他化学部分可以与其共价连接、对其进行修饰、或从其上消除,以形成多种具有共同的结构要素的分子。示例性的部分包括卤素原子、羟基基团、甲基基团、硝基基团、羰基基团或任何其他类型的分子基团。骨架的特征可包括,例如,在靶分子结合位点处结合,使得骨架上的一个或多个取代基位于靶分子结合位点中的结合口袋中;具有可以被化学修饰(特别是通过合成反应)的化学易处理结构,使得可以容易地构建组合文库;具有在此处可连接有部分而不干扰骨架与蛋白质结合位点的结合的化学位置,使得骨架或文库成员可被修饰以形成配体,以实现其他期望的特性,例如,使配体能够被主动运输到细胞和/或特定的器官内,或使得配体能够附着于色谱柱以用于其他分析。因此,分子骨架在经修饰以提高体内或体外药理学性质(例如,结合亲和性和/或特异性)之前可以是经鉴别的、小的靶标结合分子。
在小分子药物的情况下,“骨架核心”可以指其上可连接不同的取代基的分子骨架的核心结构。因此,对于特定化学品类别的多种骨架分子,骨架核心是所有骨架分子所共有的。在许多情况下,骨架核心可由一个或多个环结构组成,或包括一个或多个环结构。骨架组是指共享骨架核心并因此全部均可认为是一种骨架分子的衍生物的一组化合物。
在生物制品的情况下,“骨架”可以指一组(例如,最小的一组)氨基酸残基或核苷酸,其使得生物药物与另一种分子(例如,蛋白质、DNA、RNA、金属、肽、多糖)相互作用。例如,骨架可包含具有基序所固有的特定几何形状的氨基酸残基或核苷酸。该几何形状包括氨基酸残基之间的距离、氨基酸残基之间的夹角和氨基酸残基的取向。
在生物制品的情况下,“骨架核心”可以指所有骨架分子所共有的一组共有氨基酸残基或核苷酸。可对骨架核心进行取代,如突变、缺失、插入。因此,对于特定生物制品类别的多种骨架分子,骨架核心是所有骨架分子所共有的。骨架组是指共享骨架核心并因此全部均可认为是一种骨架分子的衍生物的一组生物制品。骨架组内的生物制品分子可共享20%、30%、40%、50%、60%、70%、80%、85%、90%、95%、98%、99%或更高的序列同源性。
骨架或骨架核心可靶向靶生物分子中的结合位点。该结合位点可以是活性结合位点或别构结合位点。通常来说,结合位点是靶分子中可与配体结合(共价地或非共价地)的区域。结合位点表现出特定的形状且可含有一个或多个结合口袋。特定形状在一类靶生物分子(如分子家族)中通常是保守的。一个类别中的结合位点也可含有保守的结构,例如,部分、结合口袋的存在和/或结合位点或结合位点的一些部分处的静电荷,所有这些均可影响结合位点的形状。结合口袋是指结合位点内的特定容积。结合口袋是结合位点内至少部分地被靶分子原子结合的特定空间。因此,结合口袋是结合位点中的特定形状、凹陷或空腔。结合口袋可含有对非共价结合另一分子很重要的特定化学基团或结构,例如,有助于分子间的离子结合、氢键结合、范德华力或疏水相互作用的基团。
本文提供的方法可用于筛选任何候选药物,包括小分子和生物制品(例如,蛋白质、肽、核酸、抗体)。例如,该方法对于筛选靶向α突触核蛋白的小分子或抗体药物是有用的。在一些方法中,鉴别出先导化合物或先导生物制品。可以根据该先导化合物或先导生物制品来设计和选择一系列的候选化合物或生物制品。例如,可使用计算机建模方法来生成候选化合物或生物制品。
在一些情况下,候选化合物或生物制品可以停靠到靶生物分子的计算机生成的结合口袋中。在一些计算机建模方法中,候选物可叠加到靶生物分子的已知先导化合物或生物制品的实验确定的排列(disposition),例如,靶生物分子-激动剂或靶生物分子-拮抗剂共晶的晶体结构中。分子优化技术可用于预测候选物与靶生物分子(或其片段)的结构匹配度,以及相互作用的能量。结构匹配度决定了候选小分子可以何种程度地物理占据结合口袋的环境中的空间;为此目的可以以多种方式构建靶生物分子(和候选物)的表面轮廓;例如,使用原子的范德华半径构建表面。相互作用的能量可通过从头计算方法(如密度泛函理论方法)或通过经验方法(如分子力学方法)来计算。相互作用的能量还可通过上述方法的结合来计算。分子优化技术还考虑到分子柔性,这表明允许键长、键角和键扭矩的变化。
靶生物分子(或靶生物分子的适当部分)的结构信息可通过X射线结晶学直接获得,通过同源性建模间接获得,或其组合。还可进行同源性建模以获得靶生物分子仅一部分的结构。这通常通过采用一种已知结构的类似部分作为模板来实现。多种方法和软件可用于此目的;例如,可以使用SWISS-MODEL中描述的方法和软件:自动化的蛋白质同源性建模服务器(Schwede,T.;Kopp,J.;Guex,N.;Peitsch,M.Nucleic Acids Research.(2003)31(13):3381-3385)。由美国专利7,384,773及其中参考文献所教导的软件和方法也可用于建模。
许多程序可用于处理机器可读的数据。例如,计算机程序DOCK及其变化形式被广泛使用(Lorber,D.和Shoichet,B.,Protein Science,7:938-950,1998)。其他示例性的程序包括Autodock(Scripps Research Institute,La Jolla,Calif.)、QUANTA(Molecular Simulations,San Diego,Calif.)、Insight(Molecular Simulations,San Diego,Calif.)、SYBYL(TRIPOS,Inc.,St.Louis,Mo.)以及LEAPFROG(TRIPOS,Inc.,St.Louis,Mo.)。
“文库”可指在本公开内容的方法中使用的一组生化实体(例如,候选生化实体)。例如,生化实体的文库可包含2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、22、24、26、28、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100、110、120、130、140、150、160、170、180、190、200、220、240、260、280、300、350、400、450、500、550、600、650、700、750、800、850、900、950、1000、1100、1200、1300、1400、1500、1600、1700、1800、1900、2000、2200、2400、2600、2800、3000、3200、3400、3600、3800、4000、4500、5000、6000、7000、8000、9000、10000种或更多种生化实体。
生化实体可以是结构上相似的。可以根据一个或多个结构相似性参数来确定两种或多种生化实体是结构上相似的,所述参数包括:分子量、化学式、总原子数、特定原子(例如,碳、氢、氮、氧和/或硫)的数目;极性;骨架核心;如通过图论测量的化学稳定性;Tanimoto(或Jaccard)系数T;芳香族基团、杂环基团、单环基团和/或稠环体系的数目;或双键的数目。例如,如果两种或多种生化实体的分子量彼此在10、20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、290、300、310、320、330、340、350、360、370、380、390、400、410、420、430、440、450、460、470、480、490、500、600、700、800、900或1000g/mol以内,则可确定所述两种或多种生化实体是结构上相似的。或者,如果两种或多种生化实体的分子量彼此在5%、10%、15%、20%、25%、30%、35%、40%、45%或50%以内,则可确定所述两种或多种生化实体是结构上相似的。在一些情况下,如果两种或多种生化实体的各种的化学式彼此在1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个原子以内,则可确定所述两种或多种生化实体是结构上相似的。在一些情况下,如果两种或多种生化实体的各种之间的原子总数之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30,则可确定所述两种或多种生化实体是结构上相似的。在进一步的情况下,如果两种或多种生化实体的各种之间的一种或多种特定原子(例如,碳、氢、氮、氧和/或硫)数目之差小于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20,则可确定所述两种或多种生化实体是结构上相似的。例如,如果碳原子数之差小于5和/或氧原子数之差小于3,则可确定所述两种或多种生化实体是结构上相似的。在一些情况下,如果两种或多种生化实体的极性[例如,如通过log D、log P(分配系数)或PSA(极性表面积)测量的]彼此在1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、12%、12%、14%、16%、18%、20%、25%、30%、35%、40%、45%或50%以内,则可确定所述两种或多种生化实体是结构上相似的。在一些情况下,如果两种或多种生化实体具有相同的骨架核心(如上文定义的),则可确定所述两种或多种生化实体是结构上相似的。在其他情况下,如果两种或多种生化实体表现出如通过图论测量的化学相似性,则可确定所述两种或多种生化实体是结构上相似的。在进一步的情况下,如果两种或多种生化实体表现出大于0.50、0.55、0.60、0.65、0.70、0.75、0.80、0.81、0.82、0.83、0.84、0.85、0.86、0.87、0.88、0.89、0.90、0.91、0.92、0.93、0.94、0.95、0.96、0.97、0.98或0.99的Tanimoto(或Jaccard)系数T,则可确定所述两种或多种生化实体是结构上相似的。在一些情况下,如果两种或多种生化实体的各种的芳香族基团、杂环基团、单环基团和/或稠环体系的数目之差小于1、2、3、4、5、6、7、8、9或10,则可确定所述两种或多种生化实体是结构上相似的。在一些情况下,如果两种或多种生化实体的各种的双键数目之差小于1、2、3、4、5、6、7、8、9或10,则可确定所述两种或多种生化实体是结构上相似的。
此外,可通过化学分析软件确定两种或多种生化实体。化学分析软件的实例包括但不限于Spinitex、Toxmatch和Simfinder。
B.选择候选药物
可采用本公开内容的方法来筛选候选药物,例如,具有相同或相似骨架核心的候选药物。这些方法均基于采用表面选择性技术由SHG部分、SFG部分或DFG部分(例如,附接于候选物、靶标或两者)产生的信号。该信号在靶标与候选分子结合时产生和检测。
在本发明中令人惊讶地发现,尽管共享相同或相似骨架的不同候选药物之间具有结构相似性,但这些候选药物可产生具有不同程度差异的信号。在一些情况下,如在下文实施例中详述的,两种结构上紧密相关的候选物可产生明显不同的信号,而两种结构上不相似的分子可产生非常相似的信号。更令人惊讶的是,在本发明中发现:检测到的信号差异与体内或体外药理学性质的差异相关联。因此,本发明的方法可用于筛选具有一种或多种期望的体内或体外药理学性质的化合物和生物制品。可根据本发明的各个实施方案来筛选候选药物。
1.直接比较
在一些实例中,对从两种或多种分子产生的信号进行相互比较。可在来自化学文库的候选药物或由例如本文所述的计算机建模方法产生并选择的候选药物之间进行比较。还可在候选药物与参考分子之间进行比较。参考分子可以是已知的药物,或先导化合物,或与体内或体外药理学性质(例如,期望的性质)相关的分子。参考分子与候选药物可具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。
在一些方法中,靶生物分子平行地与两种或多种候选物(例如,具有相同或相似的、靶向靶生物分子中结合位点的骨架核心的两种候选物)进行接触。在靶生物分子与各种候选物结合时产生信号。对由不同候选物产生的信号进行定量并相互比较。可以产生由不同候选物产生的信号之间的差异。该差异可以是正差异或负差异。在一些情况下,正差异与体内或体外药理学性质的改善相关。在一些情况下,正差异与体内或体外药理学性质的降低相关。在其他情况下,负差异与体内或体外药理学性质的改善相关。在一些其他情况下,负差异与体内或体外药理学性质的降低相关。
因为差异指示体内或体外药理学性质的差别,所有可以根据这一差异选择候选药物。例如,在正差异与体内或体外药理学性质的改善相关的情况下,选择具有较高信号的候选物。在负差异与体内或体外药理学性质的改善相关的情况下,选择具有较低信号的候选物。
同样地,可以简单地通过将由不同候选药物产生的信号进行排序来选择候选药物。在信号增高与体内或体外药理学性质的改善相关的情况下,选择具有较高信号的候选物。在信号降低与体内或体外药理学性质的改善相关的情况下,选择具有较低信号的候选物。
不同候选物和参考分子的信号可以是浓度归一化的。在一些方法中,噪声水平以上1%、5%、10%、20%、30%、40%、50%、100%、200%、300%、400%、500%的信号差异指示体内或体外药理学性质的差异。
2.与候选分子预温育
在一些实例中,间接地比较来自候选药物和参考分子的信号。参考分子可以是已知的药物,或先导化合物,或与体内或体外药理学性质(例如,期望的性质)相关的分子。参考分子可以与候选分子靶向相同的结合位点。参考分子与候选药物可具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。
具体而言,靶生物分子首先与候选药物接触,形成靶生物分子与候选分子的结合复合物。然后该结合复合物与参考分子接触。如下文实施例中详述的,结合复合物与参考分子接触时产生的信号与分子的体内或体外药理学性质相关。如果候选分子具有与参考分子相比降低的体内或体外药理学性质,则可在结合复合物与参考分子结合时检测到信号。如果候选分子具有与参考分子相比提高的体内或体外药理学性质,则可能检测不到信号或仅检测到很小的信号。因此,可基于这一相关性来筛选和选择候选分子,即,无信号或小的信号指示与参考分子相比增加或至少相当的体内或体外药理学性质。
不同候选物和参考分子的信号可以是浓度归一化的。该信号可以是正信号或负信号。在一些方法中,高于噪声水平不超过1%、5%、10%、20%、30%、40%、50%、100%或200%的信号指示与参考分子相比增加的或至少相当的体内或体外药理学性质。在一些实例中,使用不超过1%、5%或10%的信号作为截断值,以用于选择或鉴别具有与参考分子相比增加的或至少相当的体内或体外药理学性质的候选物。
3.与参考分子预温育
在一些实例中,间接地比较来自候选药物和参考分子的信号。参考分子可以是已知的药物,或先导化合物,或与体内或体外药理学性质(例如,期望的性质)相关的分子。参考分子可以与候选分子靶向相同的结合位点。参考分子与候选药物可具有相同或相似的骨架核心,该骨架核心靶向靶生物分子中的结合位点。
具体而言,靶生物分子首先与参考候选物接触,形成靶生物分子与参考分子的结合复合物。然后该结合复合物与候选分子接触。如果候选分子具有与参考分子相比降低的体内或体外药理学性质,则可能检测不到信号或仅检测到很小的信号。如果候选分子具有与参考分子相比提高的体内或体外药理学性质,则可在结合复合物与参考分子结合时检测到信号。因此,可基于这一相关性来筛选和选择候选分子,即,信号指示与参考分子相比增加的或至少相当的体内或体外药理学性质。
不同候选物和参考分子的信号可以是浓度归一化的。该信号可以是正信号或负信号。在一些方法中,高于噪声水平超过1%、5%、10%、20%、30%、40%、50%、100%或200%的信号指示与参考分子相比增加的或至少相当的体内或体外药理学性质。在一些实例中,使用超过1%、5%或10%的信号作为截断值,以用于选择或鉴别具有与参考分子相比增加的或至少相当的体内或体外药理学性质的候选物。
A.其他测定
一旦采用本文所述的方法鉴别了具有期望性质的候选药物,该性质可通过一种或多种体内或体外测定来确认。
体外测定可包括但不限于结晶学、核磁共振光谱法、质谱法和细胞测定。细胞测定的基本原理是本领域公知的,且包括这样一些方法,在这些方法中将候选药物递送至细胞中(例如,通过电穿孔、被动扩散、显微注射等)并且测量活性终点,例如已知受被测量的特定候选药物的活性影响的细胞表型变化。
体内测定包括在动物模型(例如,整个动物或动物器官)中筛选候选药物。因此,可以进一步在细胞模型或动物模型中对候选药物引起与特定活性相关的表型的可检测变化的能力进行检测。除细胞培养之外,还可采用动物模型来检测候选药物治疗动物模型中的疾病的能力。
B.体内或体外药理学性质
如上所述,所检测到的信号或信号差异可用于确定体内或体外药理学性质。在一些情况下,该药理学性质可包括药代动力学性质。可采用本文所述的方法筛选的相关体外药理学性质包括但不限于结合亲和性、结合特异性和结合亲合性。可采用本文所述的方法筛选的相关体内药理学性质包括但不限于治疗效力、药代动力学性质、临床前毒性性质、临床效力性质、临床毒性性质以及临床安全性性质。
检测的信号或信号差异可用于确定功能效果。例如,检测到的信号或信号差异可以与功能表型(例如,体内的)、作用机制(例如,体内或体外的)或其组合相关。在一些实例中,检测到的信号的差异可以由候选药物产生的信号与已知药物产生的信号的比较造成,其中候选药物的功能效果(例如,功能表型、作用机制)可根据所检测到的信号(例如,SHG信号特征、SFG信号特征或DFG信号特征)与已知药物的信号的相似性来确定。在一些情况下,体外和/或体内药理学性质可以与功能效果关联或相关,反之亦然。例如,治疗效力、结合亲和性或任何其他体外或体内药理学性质可以与采用本文所述的方法确定的作用机制相关联。因此,本公开内容中针对筛选体外或体内药理学性质而描述的任何方面均可至少在一些配置下同等地应用于筛选体外或体内的功能表型和/或作用机制。
治疗效力是指药物或候选药物在受试者(例如,人类、动物)中的治疗效果。治疗效力是药物或候选药物的以下能力:(a)通过减少疾病状态或病理学状况发作的可能性或使其延迟,从而防止疾病状态或病理学状况的发展,或(b)减少或消除一些或全部与疾病状态或病理学状况相关的临床症状。
相关药代动力学性质包括生物利用度(即,作为完整药物到达全身循环的剂量的分数)、未结合的分数(即,药物在血浆或血液中结合的程度;[未结合的药物浓度]/[总药物浓度])、被药物代谢酶代谢的药物的分数、血浆药物浓度-时间曲线下总面积以及单位时间清除药物的血液的体积。
相关临床前毒性性质包括50%治疗剂量(TD50;导致50%的测试动物达到期望的治疗结果的药物剂量)、50%致死剂量(LD50;导致50%的测试动物死亡的药物剂量)以及治疗指数(LD50/TD50之比;药物固有毒性的量度)。
相关临床效力性质包括需要治疗以达到1种期望结果的患者数,以及比值比(odds)(与对照中没有达到期望结果的几率(odds)相比,在治疗的病例中达到期望结果的几率)。
相关临床毒性性质包括需要治疗以具有1种毒性结果的患者数,以及比值比(与对照中没有达到药物毒性的几率相比,在治疗的病例中达到药物毒性的几率)。
在一些情况下,检测到的信号或信号差异不仅与体外药理学性质相关,而且与体内药理学性质相关。在一些情况下,检测到的信号或信号差异与体内药理学性质相关,但不与体外药理学性质相关。在一些情况下,检测到的信号或信号差异与体外药理学性质相关,但不与体内药理学性质相关。
在一些情况下,检测到的信号或信号差异与超过一种体内或体外药理学性质相关。例如,在一些情况下,检测到的信号或信号差异不仅与结合亲和性相关,而且与其他性质如治疗效力、药代动力学性质、临床前毒性、临床效力或临床毒性相关。
在一些情况下,检测到的信号或信号差异与一些体内或体外药理学性质的差异相关,但不与其他体内或体外药理学性质的差异相关。例如,在一些情况下,检测到的信号或信号差异与治疗效力、药代动力学性质、临床前毒性、临床效力或临床毒性相关,但不与结合亲和性、结合特异性或结合亲合性相关。
II.鉴别针对靶生物分子的结合部分的方法
本文提供了用于鉴别针对靶生物分子的结合部分(例如,与体内或体外药理学性质相关的结合部分)的方法。该方法基于采用表面选择性技术由SHG活性部分、SFG活性部分或DFG活性部分(例如,附接于候选物或靶标)产生的信号。通常,该方法包括比较两种或多种候选物(例如,具有相同或相似骨架核心的候选物)的结构。该鉴别是基于靶标与不同候选物结合时产生并检测的信号之间的信号差异。
如上所述,令人惊讶地发现:所述信号差异与体内或体外药理学性质的差异相关联。因此,采用本文所述的方法鉴别的结合部分不仅仅只是另外的结合部分。相反,所鉴别的结合部分是功能上相关的部分,其增强候选物的一种或多种期望的体内或体外药理学性质。
在一些情况下,所鉴别的结合部分不仅与体外药理学性质相关,而且与体内药理学性质相关。在一些情况下,所鉴别的结合部分与体内药理学性质相关,但不与体外药理学性质相关。在一些情况下,所鉴别的结合部分与体外药理学性质相关,但不与体内药理学性质相关。
在一些情况下,所鉴别的结合部分与超过一种体内或体外药理学性质相关。例如,在一些情况下,所鉴别的结合部分不仅与结合亲和性相关,而且与其他性质如治疗效力、药代动力学性质、临床前毒性、临床效力或临床毒性相关。
在一些情况下,所鉴别的结合部分与一些体内或体外药理学性质的差异相关,但不与其他体内或体外药理学性质的差异相关。例如,在一些情况下,所鉴别的结合部分与治疗效力、药代动力学性质、临床前毒性、临床效力或临床毒性相关,但不与结合亲和性、结合特异性或结合亲合性相关。
可根据以上针对筛选靶向靶生物分子的候选药物的方法所述的多种方法来测量分子间的信号差异。例如,可通过以下方法测量分子间的信号差异:直接将两种或多种分子产生的信号进行比较(第I.B.1部分),检测靶生物分子和候选分子的结合复合物与参考分子接触时的信号(第I.B.2部分),或检测靶生物分子和参考分子的结合复合物与候选分子接触时的信号(第I.B.3部分)。
III.用于确定结构-活性关系和/或对生化实体进行编目的方法
本文提供了用于确定结构-活性关系和/或对生化实体或其骨架进行编目的方法。例如,该结构-活性关系可以采用候选生化实体的信号特征(例如,SHG、SFG、DFG)来确定。在一些情况下,候选生化实体的信号特征可以与检测的另一种生化实体的信号特征相比较。检测的信号特征的相似性可用于关联药学性质或功能效果。
例如,信号特征可包括大小、方向或单位时间的信号。信号特征还可包括与基线相比大小或方向的变化,或动力学的变化。可以在接触候选生化实体之前,且任选地在与物剂(例如,另一种候选生化实体)温育之后,测量基线。而且,信号特征可通过测量检测信号的动力学(实时)或终点变化来产生。
其他生化实体可以是具有一种或多种药理学性质或功能效果(例如,结合性质、功能性质、效力、效能、选择性、以上的任意组合)的已知构象调节剂。在这类情况下,如果候选生化实体产生与已知构象调节剂相似的信号特征,则该候选生化实体可被确定为也具有一种或多种已知的药理学性质或功能效果。而且,如果候选生化实体与已知的构象调节剂具有一个或多个共同的结构参数[例如,骨架、骨架核心、分子量、电荷、化学式、总原子数、特定原子(例如,碳、氢、氮、氧、硫)的数目、极性(例如,如通过log D、log P、PSA测量的)、如通过图论测量的化学相似性、Tanimoto(或Jaccard)系数T、芳香族基团的数目、杂环基团的数目、单环基团的数目、稠环体系的数目以及双键的数目],则这一个或多个结构参数也可被确定为有助于一种或多种已知的药理学性质或功能效果。
或者,其他生化实体可以是另一种候选生化实体。如果候选生化实体产生相似的信号特征(例如,SHG信号特征),则该信号特征可用于将该候选生化实体与共同的药理学性质或功能效果相关联。这可以应用于候选生化实体的整个文库,其中可根据该信号特征对候选生化实体进行分组或“编目”。在一些实例中,具有相似信号特征的候选生化实体还可被确定为具有共同的药理学性质或功能效果。
IV.其他应用
SHG技术提供了在溶液中具有X射线水平灵敏度的用来检测构象变化的实时手段,并因此使得靶标能够对适用的全部构象进行遍历(sample)。SHG技术对药物发现可能具有重大影响。结构信息在药物发现中是高度有信息性且有验证性的,并且通常在生化试验或基于细胞的试验之后进行。这些试验通常产生许多命中,在通过结构技术进行更加严格的检查时,这些命中被证明是假阳性。采用NMR或X射线结晶学来鉴别(例如,挑出)真正的命中(例如,真阳性)是费时且通常不切实际的(Keseru GM和Makara GM,Drug Discovery Today 11(15-16):741-748,2006)。我们的方法将生化筛选(即,基于分子的试验)与动力学的、结构读出相结合。实际上,SHG提供了一种研究结构动力学和构象变化的新技术,结构动力学和构象变化与生物功能是同义的。在该技术的第一次展示中,我们鉴别了能够阻断精胺对α-突触核蛋白的作用的单个配体,且通过NMR确认它自身似乎起着类似于精胺的作用;这本身就是令人惊讶且不寻常的结果,因为通常在初级筛选指示的化合物为命中与随后通过结构技术的确认之间相距甚远。而且,我们针对这样一种靶标鉴定该配体,对于该靶标有较少的已知结合物并且没有其他针对C末端区域的结合物。
SHG筛选可以以多种模式使用。例如,可以筛选抑制已知配体诱导的构象变化的配体(例如,拮抗剂)或在一个或多个感兴趣的位点独自诱导构象变化的配体(例如,激动剂)。所寻找的配体的类型可包括用于治疗学的先导化合物、别构调节剂以及用于化学生物学和疾病发病机理研究的工具化合物(tool compound),等等。如果特定的试验涉及已知的工具化合物,则SHG可用于提供关于其作用机制以及其结合亲和性的信息。尽管大多数筛选文库的大部分均含有发荧光且可干扰荧光测定中的读出的化合物,但极少部分有可能是SH活性的,这是更严格的一组理化性质。
在溶液中实时检测构象变化有可能揭示出在X射线实验中观察不到的新的结合位点,如别构结合位点。该技术作为一种鉴别目前被认为是“不可用作药物的”靶标如Ras的别构调节剂的手段有很大的前途。由于该技术的结构敏感性,即使是在构象空间中在正确方向上产生变化的弱结合剂也可得到快速地鉴别和优化。通过这一方式,该技术提供了一种鉴别无法以其他任何方式鉴别的药物新类型以及开发具有精确定制的构象(因而,功能)效果的药物的途径。
蛋白质构象变化是大多数生物过程中的主要功能事件。其研究方法通常依赖于检测由分子环境的变化引起的信号变化,而不是其真实空间坐标的变化。SHG对真实空间的结构变化是高度且直接敏感的。如本文所述,当结构变化实时发生时且在使得蛋白质可以遍历(sample)适用的完整构象空间的生理条件下,SHG可用于位点特异性地筛选蛋白质的结构变化。这些性质使得该技术是用于鉴别调节蛋白质构象的配体(如别构药物或内源性生物调节剂)的理想技术。通过研究在多个探针位点处的构象变化,该技术可提供对构象变化的总体剖析以及对在这些特定组的位点引起结构变化的配体进行鉴别和分类的选择性手段。在一些情况下,SHG筛选平台还可对探针的角度变化(以度为单位)进行定量,使得在不同位点的测量可提供用于建立构象变化的原子解析图的信息。简而言之,该技术将在结构生物学和药物发现的无数领域中具有显著的影响。
V.信号特征
本文所述的方法可包括采用表面选择性技术(例如,SHG、SFG、DFG)来测量生化实体(例如,靶生化实体)的构象状态。在一些实施方案中,可采用表面选择性技术测量生化实体的构象状态,以产生基线(例如,SHG基线)。然后,可再次采用表面选择性技术(例如,在诸如使靶生化实体与候选生化实体接触的事件之后)测量生化实体的构象状态,以产生信号特征(例如,SHG、SFG或DFG信号特征)。该信号特征可指示生化实体的构象状态的变化。例如,当第一生化实体与第二生化实体接触(例如,结合)时,其构象状态可发生变化。该第二生化实体可经历或可不经历其构象状态的变化。在一些情况下,第一和第二生化实体均可经历构象变化(例如,两者之间的结合复合物的变化)。在一些情况下,第二生化实体可经历影响第一生化实体的构象的构象变化。
在一些实施方案中,可以不测量基线。例如,可在第一生化实体与第二生化实体接触时测量第一生化实体的第一构象状态。接下来,可在第一生化实体(例如,第一生化实体的相同拷贝,或第一生化实体的不同拷贝)与第三生化实体接触时测量第一生化实体的第二构象状态,且可通过相减或比较信号特征来观察构象状态的差异。因此,在一些情况下,信号特征可相对于基线来定义(例如,相对于基线响应的SHG、SFG或DFG响应)。在一些情况下,信号特征可以不相对于基线来定义,或者可通过减去共同的基线来定义。
采用本公开内容的表面选择性技术产生的信号特征可包括在不同时间测量的非线性光学响应(例如,SHG响应)。例如,可通过测量生化实体的构象状态的动力学(实时)变化,测量生化实体的构象状态的终点变化,或在任何其他时间(例如,在生化实体与另一生化实体接触之前、期间或之后的任何时间)或在多个预定的时间(例如,接触前、接触时、接触后的1、2、3、4、5、6、7、8、9、10个或更多个时间,或其任何组合)测量生化实体的构象状态的变化,来产生信号特征。在一些情况下,信号特征可通过测量非线性光学信号强度(例如,SHG强度)而产生。
本公开内容的信号特征可用作筛选、选择和/或评估生化实体的基础。例如,可根据其信号特征(例如,第一生化实体与第二生化实体的相互作用如结合的信号特征)从一组候选生化实体中选择生化实体。在一些情况下,该选择可包括将信号特征与一种或多种其他信号特征(例如,已知化合物的参考信号特征、相似生化实体的信号特征等)进行比较。在一个实例中,选择具有与由已知构象调节剂诱导的构象状态变化最相似的构象状态变化的生化实体。由已知构象调节剂产生的构象状态变化可与药理学性质或功能效果(例如,作用机制、表型、临床性质、结合性质等)相关。因此,在一些情况下,该比较可用于选择具有相似或基本相同的药理学性质或功能效果的生化实体。在另一个实例中,将生化实体的信号特征进行比较(例如,第一生化实体的信号特征与第二生化实体的信号特征相比较),并根据该比较来选择生化实体(例如,在生化实体组或文库中)。在另一个实例中,根据信号特征与已知药理学性质或已知功能效果的相关性来选择生化实体。
生化实体可具有(例如,彼此之间或与参考生化实体如已知构象调节剂或被批准的药物)基本相似或不同的信号特征。例如,在筛选试验中,当各种候选生化实体与相同的靶生化实体接触时,各种候选生化实体可产生不同的信号特征。
在一些情况下,本文的信号特征可以与已知的药理学性质或已知的功能效果相关。例如,在筛选试验中,候选生化实体(例如,至少两种候选生化实体)可具有相似的药理学性质或功能效果。例如,药理学性质或功能效果(例如,结合亲和性、结合特异性、临床效力、表型变化等)可以是至少约1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或99%相似的,等等。在另一个实例中,在筛选试验中,候选生化实体(例如,至少两种候选生化实体)可不具有相似的药理学性质或功能效果。例如,药理学性质或功能效果(例如,结合亲和性、结合特异性、临床效力、表型变化等)可以是至多约1%、5%、10%、20%、30%、40%、50%、60%、70%、80%、90%、95%或99%相似的,等等。在一些情况下,一种或多种药理学性质或功能效果的相似性可以与相应信号特征的相似性相关。在其他情况下,一种或多种药理学性质或功能效果的相似性可以与相应信号特征的相似性不相关。
本公开内容的信号特征的产生可包括测量一种或多种非线性光学(例如,SHG)参数。例如,这些参数可包括非线性光学(例如,SHG)信号的大小相对于非线性光学(例如,SHG)基线的变化、非线性光学(例如,SHG)信号的方向相对于非线性光学(例如,SHG)基线的变化、接触时单位时间非线性光学(例如,SHG)信号率的变化,或其任意组合。可采用计算机可执行程序分析信号特征。在一些情况下,可分析信号特征的相似性。在一些情况下,相似的信号特征可作为选择(或不选择)生化实体的依据在本公开内容的筛选方法中使用。在一些情况下,相似的信号特征可作为关联或分析SAR的依据在本公开内容的评估方法中使用(例如,产生一组生化实体的结构-活性关系(SAR))。
在一些情况下,相似性可采用非线性光学(例如,SHG)参数来定义。例如,如采用一个或多个非线性光学(例如,SHG)相似性参数确定的(例如,下列相似性参数的任意组合),由第一生化实体产生的信号特征和由第二生化实体产生的信号特征可以是相似的。在一些实例中,该相似性参数可以是:第一候选生化实体与靶生化实体接触时非线性光学(例如,SHG)信号的大小相对于非线性光学(例如,SHG)基线的变化与第二候选生化实体与靶生化实体接触时非线性光学(例如,SHG)信号的大小相对于非线性光学(例如,SHG)基线的变化相差小于约1%、5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、90%或更多以内(例如,约50%以内)。在一些实例中,该相似性参数可以是:第一候选生化实体与靶生化实体接触时非线性光学(例如,SHG)信号的方向相对于非线性光学(例如,SHG)基线的变化和第二候选生化实体与靶生化实体接触时非线性光学(例如,SHG)信号的方向相对于非线性光学(例如,SHG)基线的变化相同。在一些实例中,该相似性参数可以是:第一候选生化实体与靶生化实体接触时单位时间非线性光学(例如,SHG)信号率的变化与第二候选生化实体与靶生化实体接触时单位时间非线性光学(例如,SHG)信号率的变化相差小于约1.1、1.2、1.3、1.4、1.5、1.6、1.7、1.8、1.9、2、2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9、3、3.5、4、4.5、5、6、7、8、9、10、15、20倍或更多倍之内(例如,约两倍),或反之亦然。
在一些实施方案中,信号特征可包括非线性光学(例如,SHG)信号响应的特有形状或轮廓。例如,信号特征可具有给定数量的特征,例如,局部极大值、局部极小值、拐点、整体斜率、斜率变化等。在一些情况下,这些特性或特征可相对于非线性光学(例如,SHG)基线而测量。在一些情况下,该相似性参数可以是:第一候选生化实体与靶生化实体接触时测量的非线性光学(例如,SHG)信号和第二候选生化实体与靶生化实体接触时测量的非线性光学(例如,SHG)信号共享至少1、2、3、4、5、6、7、8、9、10、15、20、30、40、50种或更多种这类特征(例如,局部极大值、局部极小值、拐点、整体斜率和/或斜率变化)。在一些情况下,该相似性参数可以是:在第一候选生化实体与靶生化实体接触时测量的非线性光学(例如,SHG)信号的局部极大值、局部极小值、拐点、整体斜率、斜率变化和/或其他信号特征中的一个或多个数量与在第二候选生化实体与靶生化实体接触时测量的非线性光学信号的局部极大值、局部极小值、拐点、整体斜率、斜率变化和/或其他信号特征的数量相同。
VI.靶生物分子和参考生化实体
供本文所述的任何方法使用的靶生物分子可以是任何生物分子,例如,蛋白质、DNA、RNA或多糖。靶生物分子可以是来自任何天然来源的天然存在的物质,或其亚单位或结构域,包括病毒、微生物(包括细菌、真菌、藻类和原生动物)、无脊椎动物(包括昆虫和蠕虫)、正常的或病理性的动物细胞、脊椎动物(例如,哺乳动物、禽类或鱼类,以及在哺乳动物中,例如人、猿、猴、牛、猪、山羊、羊驼、绵羊、大鼠、小鼠、兔、豚鼠、猫和狗)或者正常的或病理性的植物细胞。或者,靶生物分子可以是已经在体外产生或者经修饰(例如通过突变、嵌合蛋白或人工蛋白质)的非天然存在的生物分子。
例如,靶蛋白可以是糖蛋白、脂蛋白、磷蛋白或金属蛋白。靶蛋白可以是核蛋白质、细胞质蛋白质、膜相关蛋白质或者分泌的蛋白质。靶蛋白并不需要是单个大分子。例如,靶蛋白可以是大分子的同质或异质多聚体(例如但不限于二聚体、三聚体或四聚体)。此外,靶蛋白可能需要一个或多个配体来行使生理功能,例如其他蛋白质、寡肽或多肽、核酸、糖、脂质或有机或无机小分子或离子。另外的实例包括辅因子、核糖体、多核糖体和染色质。
靶蛋白包括但不限于固有无序蛋白质(IDP),例如,α突触核蛋白(其隐含于帕金森病)和COW(其隐含于阿尔茨海默病)。靶蛋白的进一步实例包括但不限于核受体、孤儿核受体、酪氨酸激酶受体、内皮素、促红细胞生成素受体、FAS配体受体、蛋白激酶(例如,蛋白激酶C、酪氨酸激酶、丝氨酸激酶、苏氨酸激酶、核苷酸激酶或多核苷酸激酶)、蛋白磷酸酶(丝氨酸/苏氨酸磷酸酶、酪氨酸磷酸酶、核苷酸磷酸酶、酸性磷酸酶、碱性磷酸酶或焦磷酸酶)、细胞周期调节物(细胞周期蛋白cdk2、CDC2、CDC25、P53、RB)、GTP酶、Rac、Rho、Rab、Ras、内切蛋白酶、外切蛋白酶(exoprotease)、金属蛋白酶、丝氨酸蛋白酶、半胱氨酸蛋白酶、核酸酶、聚合酶、逆转录酶、整合酶、离子通道、陪伴蛋白(例如,热休克蛋白)、脱氨酶、核酸酶(例如,脱氧核糖核酸酶、核糖核酸酶、内切核酸酶、外切核酸酶)、端粒酶、引发酶、解旋酶、脱氢酶、转移酶(肽基转移酶、转氨酶、糖基转移酶、核糖基转移酶(ribosyltransferases)、乙酰基转移酶、鸟苷酸转移酶或甲基转移酶)、水解酶、羧化酶、异构酶、糖苷酶、脱氨酶、脂肪酶、酯酶、硫酸酯酶、纤维素酶、裂合酶、还原酶、连接酶或细胞泛素化途径的加工酶(如E1、E2或E3酶或去泛素化酶)。在一些实施方案中,供本文公开的任何方法使用的靶蛋白可以是选自病毒蛋白质、细菌蛋白质、植物蛋白质、动物蛋白质和人蛋白质的结构和非结构蛋白质。在一些实施方案中,靶蛋白可以是病毒蛋白质,例如但不限于流感病毒、甲型肝炎病毒、乙型肝炎病毒、丙型肝炎病毒、人免疫缺陷病毒、禽流感病毒、埃博拉(Ebola)病毒、SARS病毒、汉坦病毒或东方马脑炎病毒的蛋白质。
靶蛋白可以是受体,包括表面和细胞内受体。在一些实施方案中,靶蛋白是核受体。核受体是一类配体激活的转录激活物。这些受体组织成用于配体结合、二聚化、反式激活和DNA结合的不同的结构域。类固醇受体家族是包含糖皮质激素、盐皮质激素、雄激素、孕激素和雌激素的受体的大家族。当配体结合时发生受体激活,其诱导构象变化,从而允许受体二聚化和辅激活蛋白的结合。这些辅激活物转而又促进受体与DNA的结合以及后续的靶基因的转录激活。除了辅激活蛋白的募集之外,配体的结合也被认为将受体置于替换或阻止作为受体功能的协阻抑物的蛋白质的结合的构象中。如果配体是药理学激动剂,则新的构象是与生物信号转导途径的其他组分例如转录因子相互作用从而在靶组织中引起生物响应的构象。如果配体是药理学拮抗剂,则新的构象是其中受体不能被一种或多种激动剂激活(在其他情况下,该激动剂能够激活该受体)的构象。NR的非详尽清单描述于国际专利申请公开号2006/046134中,该申请的公开内容通过引用并入本文(参见第14和15页以及图1)。在一些实施方案中,供本文公开的任何方法使用的NR可选自雌激素受体、雄激素受体、糖皮质激素受体、视黄酸受体α(RARG)、视黄酸X受体(RXR)、过氧化物酶体增殖物激活受体(PPAR)、肝X受体α(LXRG)或孕酮受体。
靶蛋白可以是G蛋白偶联受体(也称为七跨膜域受体),即,通过与G蛋白偶联而介导信号转导的受体的超家族的任何成员。GPCR包括跨膜受体蛋白的大家族(占人类总基因组的约5%),这些蛋白质与在细胞外环境中存在的分子相结合并且能够在细胞内触发信号转导级联并最终引发细胞应答。GPCR仅发现于包括酵母、领鞭虫类和动物在内的真核生物中。结合并激活这些受体的分子包括但不限于光敏化合物、气味、信息素、激素以及神经递质,并且其大小从小分子到肽直至大的蛋白质不等。影响胞质钙水平的一类GPCR的一个实例通过Gq类型的G蛋白发挥作用,该Gq类型的G蛋白激活磷脂酶C(PLC)途径,导致磷酸肌醇的水解以产生两类不同的第二信使,即,二酰基甘油和肌醇磷酸。二酰基甘油转而激活某些蛋白激酶C(PKC),而肌醇磷酸(例如但不限于IP3)刺激钙从细胞内储库(store)如内质网、肌浆网(对于肌细胞)和/或线粒体的动员。结合并激活这些受体的分子包括但不限于光敏化合物、气味、信息素、激素以及神经递质,并且其大小从小分子到肽直至大的蛋白质不等。
供本文公开的方法使用的GPCR包括但不限于:Gq蛋白或Gq/11、α-1肾上腺素能受体(α1-AR)、硬骨鱼紧张肽(UT)受体、5-HT2和5-HT6血清素受体、下丘泌素(食欲肽)受体、组胺HI受体、缓激肽B1和B2受体、铃蟾肽BB2受体、P2Y嘌呤能受体、乙酰胆碱受体(例如,M1、M3和M5)、mGluR5谷氨酸受体、加压素V2和VI受体、血管紧张素AGTR1受体、胆囊收缩素CCKAR和CCKBR受体、内皮素ENDRA受体、生长素释放肽GHSRla受体、褪黑激素MTNR1A受体、神经降压肽NTSR1受体、血小板活化因子PTAFR受体、促黄体激素受体(LHR)、促卵泡激素受体(FSHR)、促性腺激素释放激素受体(GnRHR)和催乳素释放肽受体PRLHR受体。在一些实施方案中,GPCR在表达钙传感蛋白的细胞中内源性表达。在其他实施方案中,GPCR在表达钙传感蛋白的细胞中异源性表达。
靶蛋白可以是激酶。激酶是将磷酸基团从高能供体分子如ATP转移到特异性底物的一类酶,这一过程被称为磷酸化。最大的激酶群体之一是蛋白激酶,其作用于并改变特定蛋白质的活性。激酶广泛地用于传送信号并控制细胞中的复杂过程。在人类中已经鉴别了超过500种不同的激酶。它们的庞大的多样性及其在信号传导中的作用使得它们成为尤其针对以异常激酶表达或调节为特征的疾病状态的研究的对象。
蛋白激酶含有大的柔性环,称为激活环或A-环,认为其构象调节激酶活性。在许多激酶中,A-环的构象由该区域内的特定残基的磷酸化来控制(Johnson 1996)。激活环通常开始于保守的AspPheGly序列并终止于保守的AlaProGlu。在非活性激酶的结构中,该环通常阻断底物或ATP结合位点(Hubbard 1994;Mohammadi 1996;和McTigue 1999)。酪氨酸激酶通常在环内具有一个或两个酪氨酸,MAPK激酶具有在T和Y上都被磷酸化的T[DE]Y基序,而大多数其他激酶在环内具有苏氨酸。
供本发明的方法使用的靶蛋白广泛地适用于任何蛋白激酶。其可以包括蛋白质酪氨酸激酶和蛋白质丝氨酸激酶。蛋白质酪氨酸激酶的非限制性实例是pp60c-src、p56lck、ZAP激酶、血小板衍生生长因子受体酪氨酸激酶、Bcr-Abl、VEGF(血管内皮生长因子)受体酪氨酸激酶和表皮生长因子受体酪氨酸激酶和表皮生长因子受体样酪氨酸激酶。适用于本发明的丝氨酸蛋白激酶的非限制性实例包括MAP(促分裂原活化蛋白)激酶、蛋白激酶C、蛋白激酶A、Akt和CDK(细胞周期蛋白依赖性蛋白激酶)。在哺乳动物生物学中,属于促分裂原活化蛋白激酶(MAPK)家族的蛋白激酶在多种与ras基因的突变和生长因子受体的失调相关的增殖性细胞疾病(例如,癌症)中被不当地激活(Magnuson等,Seminars in Cancer Biology,5:247-252(1994))。MAP激酶在本领域中是已知的,并且此类激酶的部分非限制性清单包括abl、Aurora-A、Aurora-B、Aurora-C、ATK、bcr-abl、Blk、Brk、Btk、c-Kit、c-Met、c-Src、CDK1、CDK2、CDK4、CDK6、cRafl、CSF1R、CSK、EGFR、ErbB2、ErbB3、ErbB4、ERK、Fak、fes、FGFR1、FGFR2、FGFR3、FGFR4、FGFR5、Fgr、FLK-4、Flt-1、Fms、Fps、Frk、Fyn、Hck、IGF-1R、INS-R、Jak、KDR、Lck、Lyn、MEK、p38、PDGFR、PIK、PKC、PYK2、Ros、Tiel、Tie2、Trk、Yes和Zap70。在本文所述方法的一些实施方案中,所述激酶是abl激酶。
对于激酶,已知几种类型的化合物能调节激酶的功能。例如,I型激酶抑制剂识别激酶的活性构象。它们通过呈现模拟由ATP通常形成的氢键的一至三个氢键而与ATP结合位点结合。不受限于理论,据认为,与I型激酶抑制剂相反,II型激酶抑制剂识别激酶的非活性构象,并且能够通过占据直接与ATP结合位点相邻的疏水口袋而间接地与ATP竞争。该疏水口袋是由激活环的独特DFG-out构象创建的。某些II型抑制剂能够直接与ATP结合位点形成氢键,但这不是功能性所必需的(Gotink和Verheul,Angiogenesis,2010,13(1):1–14)。在另一方面,III型激酶抑制剂是非ATP竞争性激酶抑制剂,其通过与激活环以外的位点结合(即通过与激酶上的别构位点结合)来调节激酶活性。由于III型化合物与激酶上的保守性较低的位点结合,它们是高度选择性的并且在研究和药物发现团体中受到越来越多的关注。
小GTP酶的Ras家族由被核苷酸鸟苷-5'-三磷酸(GTP)调节的细胞内信号分子组成。这些蛋白质起二元信号开关的作用,具有“打开”和“关闭”两种状态。在“关闭”状态,Ras结合至核苷酸鸟苷二磷酸(GDP),而在“打开”状态,Ras结合至GTP。因此,Ras的活化和去活化受活性GTP结合形式与非活性GDP结合形式之间的循环控制。在GTP结合的构象中,Ras对许多效应物具有高亲和性,这允许执行其细胞内功能。Ras调节的信号通路控制以下多样化的过程:肌动蛋白细胞骨架完整性、增殖、分化、细胞粘附、凋亡和细胞迁移。在一些情况下,突变导致组成型Ras活化(即,无论其是否结合至GDP,均无法成为其“关闭”状态的Ras蛋白),从而导致异常的信号传导。本文的方法可用于筛选或鉴别能够特异性调节Ras行为(例如,癌细胞中)以减少下游Ras效应物分子的异常信号传导的化合物。
预计任何Ras蛋白均可用于任何本文公开的方法中。三种人类ras基因编码由188至189个氨基酸的链组成的、极其同源的蛋白质,这些蛋白质被称为H-RAS、N-RAS以及K-RAS4A和K-RAS 4B(两种K-RAS蛋白由选择性剪接产生)。尽管Ras亚家族在临床上最值得注意的成员为H-RAS、K-RAS和N-RAS(主要因为与多种类型的癌症有关),但该亚家族还存在许多其他成员,其可包括但不限于DIRAS1、DIRAS2、DIRAS3、ERAS、GEM、MRAS、NKIRAS1、NKIRAS2、NRAS、RALA、RALB、RAP1A、RAP1B、RAP2A、RAP2B、RAP2C、RASD1、RASD2、RASL10A、RASL10B、RASL11A、RASL11B、RASL12、REM1、REM2、RERG、RERGL、RRAD、RRAS或RRAS2(参见,Wennerberg等人,2005,"The Ras superfamily at a glance,"J.Cell.Sci.118(Pt 5):843–6,其公开内容通过引用全文并入本文)。本公开内容的方法的范围内使用的包括Ras蛋白的突变形式。
靶蛋白包括靶蛋白的突变形式。如本文中所用的“突变”包括在定义的一级氨基酸序列如靶蛋白的一级氨基酸序列中的至少一个氨基酸残基的氨基酸残基缺失、氨基酸残基插入和/或氨基酸残基置换。氨基酸“置换”意指定义的一级氨基酸序列的至少一个氨基酸组分被另一个氨基酸(例如,半胱氨酸残基或赖氨酸残基)替换。通过标准技术将突变或置换工程构建到靶蛋白的一级氨基酸序列内的方法在本领域中是公知的。供本文描述的方法使用的靶蛋白包括保守置换。
靶蛋白的生物学性质的实质性改变通过选择置换来完成,该置换在其对维持以下方面的作用上显著不同:(a)在置换区域中的多肽骨架的结构,例如作为片层或螺旋构象,(b)分子在靶位点处的电荷或疏水性,或(c)侧链的体积(bulk)。非保守置换将需要将这些类别之一的成员更换成另一类别的成员。
在进一步的实施方案中,供本文公开的任何方法使用的突变靶蛋白可包含一个或多个非天然存在的或修饰的氨基酸。非天然氨基酸包括但不限于高赖氨酸、高精氨酸、高丝氨酸、氮杂环丁烷羧酸、2-氨基己二酸、3-氨基己二酸、β-丙氨酸、氨基丙酸、2-氨基丁酸、4-氨基丁酸、6-氨基己酸、2-氨基庚酸、2-氨基异丁酸、3-氨基异丁酸、2-氨基庚二酸、叔丁基甘氨酸、2,4-二氨基异丁酸、锁链素、2,2'-二氨基庚二酸、2,3-二氨基丙酸、N-乙基甘氨酸、N-乙基天冬酰胺、高脯氨酸、羟赖氨酸、别羟基赖氨酸、3-羟基脯氨酸、4-羟基脯氨酸、异锁链素、别异亮氨酸、N-甲基丙氨酸、N-甲基甘氨酸、N-甲基异亮氨酸、N-甲基戊基甘氨酸、N-甲基缬氨酸、萘丙氨酸(naphthalanine)、正缬氨酸、正亮氨酸、鸟氨酸、瓜氨酸、戊基甘氨酸、六氢吡啶羧酸和硫代脯氨酸。修饰的氨基酸包括天然的和非天然的氨基酸,其被可逆地或不可逆地化学封闭(block),或在其N-末端氨基或其侧链基团上被修饰,例如,N-甲基化的D和L氨基酸,侧链官能团被化学修饰成另一个官能团。例如,修饰的氨基酸包括甲硫氨酸亚砜、甲硫氨酸砜、天冬氨酸-(β-甲酯)、天冬氨酸的修饰的氨基酸、N-乙基甘氨酸、甘氨酸的修饰的氨基酸,或丙氨酸羧酰胺,丙氨酸的修饰的氨基酸。另外的非天然和修饰的氨基酸以及将其掺入至蛋白质和肽中的方法在本领域中是已知的(参见,例如Sandberg等,(1998)J.Med.Chem.41:2481-91;Xie和Schultz(2005)Curr.Opin.Chem.Biol.9:548-554;Hodgson和Sanderson(2004)Chem.Soc.Rev.33:422-430)。
在一些实施方案中,供本文描述的方法使用的突变靶蛋白可以通过适当的纯化方案使用标准蛋白质纯化技术从细胞(如癌细胞)中分离。在另一实施方案中,供本文描述的方法使用的突变靶蛋白通过重组DNA技术产生。作为重组表达的替代,供本文所述方法使用的突变靶蛋白可以使用标准肽合成技术进行化学合成。
此外,可使参考生化实体经受本申请中描述的方法以产生参考信号特征,该信号特征可用于将候选生化实体的信号特征与药理学性质和/或功能效果相关联。例如,具有一种或多种已知药理学性质和/或功能效果的参考生化实体可具有能够与候选生化实体的文库的信号特征进行比较的参考信号特征。具有与参考信号特征相似的信号特征的候选生化实体可与一种或多种已知药理学性质和/或功能效果相关联,和/或可进一步测试其一种或多种已知药理学性质和/或功能效果。
参考生化实体可以是具有已知功能和/或已知靶标的任何已知的药物。例如,参考生化实体可以是抗癌药或抗炎药。参考生化实体还可以是已知的激酶抑制剂,包括但不限于酪氨酸激酶抑制剂和丝氨酸/苏氨酸激酶抑制剂。参考生化实体的实例包括但不限于:安律凡(Abilify)、耐信(Nexium)、修美乐(Humira)、可定(Crestor)、Advair Diskus、恩利(Enbrel)、欣百达(Cymbalta)、类克(Remicade)、培非格司亭(Neulasta)、克帕松(Copaxone)、Lantus Solostar、利妥昔单抗(Rituxan)、思力华(Spiriva)、捷诺维(Januvia)、Atripla、兰德仕(Lantus)、阿瓦斯汀(Avastin)、利痛抑(Lyrica)、奥施康定(OxyContin)、依泊汀(Epogen)、西乐葆(Celebrex)、特鲁瓦达(Truvada)、代文(Diovan)、格列卫(Gleevec)、赫赛汀(Herceptin)、雷珠单抗(Lucentis)、Vyvanse、Zetia、美金刚(Namenda)、Levemir、信必可(Symbicort)、Tecfidera、昂斯妥凝胶(AndroGel)、Novolog、依诺肝素(enoxaparin)、哌醋甲酯(methylphenidate)、赛宝松(Suboxone)、拜瑞妥(Xarelto)、ProAir HFA、优泌乐(Humalog)、力比泰(Alimta)、诺和力(Victoza)、万艾可(Viagra)、思瑞康XR(Seroquel XR)、Synagis、阿沃纳斯(Avonex)、诺维乐(Renvela)、利比(Rebif)、西力士(Cialis)、芬戈莫德(Gilenya)、内舒拿(Nasonex)、优特克单抗(Stelara)、丽眼达(Restasis)、布地奈德(budesonide)、醋氨酚/氢可酮、丙酸氟替卡松(Flovent HFA)、Lovaza、达芦那韦(Prezista)、Isentress、捷诺达(Janumet)、Procrit、强力霉素(doxycycline)、Orencia、安非他命/右旋安非他命、VESIcare、右兰索拉唑(Dexilant)、优保津(Neupogen)、利多卡因(lidocaine)、鲁尼斯塔(Lunesta)、非诺贝特(fenofibrate)、阿比特龙(Zytiga)、锐艾妥(Reyataz)、西那卡塞(Sensipar)、美托洛尔(metoprolol)、善思达(Invega Sustenna)、左甲状腺素(Synthroid)、阿沃纳斯(Avonex)、Prevnar 13、乐无喘(Xolair)、左旋甲状腺素(levothyroxine)、Benicar、Stribild、Zostavax、百达生(Pradaxa)、维多灵(Vytorin)、特敏福(Tamiflu)、Xgeva、易维特(Evista)、希罗达(Xeloda)、阿法达贝泊汀(Aranesp)、万托林HFA(Ventolin HFA)、双丙戊酸钠(divalproex sodium)、飞尼妥(Afinitor)、Betaseron、阿得拉XR(Adderall XR)以及Complera。
VII.非线性活性部分
在一些实施方案中,一种或多种非线性活性(例如,二次谐波活性)部分或标记物可连接至或并入靶生化实体(例如,生物分子)、候选生化实体(例如,候选分子)、参考生化实体(例如,分子)或其任意组合。在一些实施方案中,例如,在通过产生结构-活性关系筛选结构上相似的生化实体或评估靶生化实体的试验和方法中,非线性活性部分或标记物可连接至或并入靶生化实体、结构上相似的生化实体的文库中的一种或多种候选生化实体、具有不同分子结构的一组生化实体中的一种或多种生化实体、参考生化实体或其任意组合。例如,在第一生化实体与第二生化实体接触(例如,导致彼此相互作用,例如,结合事件)的任何给定的试验中,第一生化实体和/或第二生化实体可包含适合于采用本文所述的表面选择性技术(例如,SHG、SFG或DFG)测量构象变化的标记物。
在一些情况下,所述标记物可以是二次谐波活性部分或标记物(又称为SHG部分或SHG标记物)。在一些情况下,和频活性或差频活性部分或标记物。在一些情况下,非线性活性部分或标记物(本文中也称为“部分”或“标记物”)可以是SHG活性的、SFG活性的、DFG活性的或其组合。
在一些实施方案中,靶生化实体(例如,生物分子如蛋白质、细胞等)可在一个或多个预定的位置被标记。例如,标记物可被放置在距离生化实体(例如,蛋白质)结构之上或之内的活性位点、结合位点、别构结合位点或任何其他位点(例如,可能经历通过试验筛选的构象变化的任何位点,经历与体内或体外药理学性质相关的构象变化的任何位点,等等)给定距离处(例如,在该位点处或与之相邻)。
在一些情况下,例如,为了能够进行本公开内容的筛选和评估试验,可在靶生化实体之上或之内的相同位置处对同一靶生化实体的多个拷贝进行标记。例如,本公开内容的系统和方法可用于检测与具有非常相似结构的生化实体相接触的靶生化实体的构象状态变化的差异。进而,该构象状态变化的差异可与药理学性质或功能效果相关联。因此,相同的标记物放置可确保与靶生化实体相互作用的生化实体在相同的条件下得到评估,并且确保观察到的靶生化实体的构象状态变化的差异是由结构、药理学性质、功能效果等中的至少一种的差异造成的,而不是由不同的标记物放置造成的。
每种标记物均可在靶生化实体上具有给定位置(例如,3D坐标)。给定的标记物可具有在靶生化实体的第一拷贝之上或之内的第一位置(例如,3D坐标(x,y)1)和在靶生化实体的第二拷贝之上或之内的第二位置(例如,3D坐标(x,y)2)。在一些实例中,第一位置与第二位置之间的相对差(例如,|(x,y)1-(x,y)2|/(x,y)1或|(x,y)1-(x,y)2|/(x,y)2)可小于约75%、50%、25%、10%、5%、1%等。在一些实例中,第一位置与第二位置之间的绝对差可小于靶生化实体的直径、高度、宽度、广度或其他特性尺寸的约1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、15%、25%、50%或75%。
部分(例如,标记物)可共价地或非共价地结合至靶生物分子,以便所得的靶生物分子对二次谐波产生具敏感性且适于使用表面选择性技术在界面进行研究。然后可以通过诸如二次谐波产生或和频产生的表面选择性技术研究标记的靶生物分子。外源性部分(例如,标记物)可以预先连接到靶生物分子上,并且在进行测量前从标记的实体上分离任何未结合的或未反应的标记物。在一个实施方案中,二次谐波活性部分(例如,标记物)在体外连接到靶生物分子上。
在一些情况下,可以使用至少两个可辨别的二次谐波活性部分(例如,标记物)。然后选择连接的两个或更多个可辨别的标记物的取向以便有助于发出的相干非线性光束的明确定义的方向。两个或更多个可辨别的标记物能够在如下的试验中使用,在该试验中,使用相对于样品以一个或多个偏振方向入射的、处于一个或多个频率的多个基本光束,得到至少两个非线性光束的发射。在一个实施方案中,二次谐波活性部分(例如,标记物)包含多个单独的二次谐波活性部分(例如,标记物),其中每一个都具有非线性磁化性并且相对于彼此以固定和确定的方向结合在一起,以增加二次谐波活性部分(例如,标记物)的总非线性磁化性。
在一些情况下,二次谐波活性部分(例如,标记物)是染料。靶生物分子可用染料通过特异性标记或非特异性标记进行标记。
二次谐波活性部分可通过特异性标记,例如通过共价键或氢键连接到靶生物分子上。例如,二次谐波活性部分(例如,标记物)可以共价地或非共价地连接到待检测的靶蛋白的一级氨基酸序列中的胺基团、赖氨酸基团或巯基基团上。在一些实施方案中,二次谐波活性部分(例如,标记物)具有胺反应性琥珀酰亚胺酯、硫醇反应性马来酰亚胺或醛和/或酮反应性酰肼和羟胺。在一些实施方案中,SFG或SHG-活性染料标记物可以通过用于偶合到叠氮化物的“点击化学法”进行缀合。用于缀合物形成的点击化学法的细节在下列文献中描述:“Synthesis and Functionalization of Biomolecules via Click Chemistry,C.Schilling等人,第15章,第355-378页,于“Click Chemistry for Biotechnology and Materials Science”J.Lahann(Ed),Wiley(2009)中。
适于在本文公开的方法中用作二次谐波或和频率活性部分(例如,标记物)的染料的实例包括:马来酰亚胺标记物(例如PyMPO-MALEIMIDETM(1-(2-马来酰亚胺基乙基)-4-(5-(4-甲氧基苯基)噁唑-2-基)吡啶鎓甲磺酸盐),其在含巯基的残基(通常为半胱氨酸)上特异性标记蛋白质)、PyMPO-NHS(其特异性标记含伯胺的残基(通常为赖氨酸))、噁唑标记物(例如特异性标记胺的PyMPO-琥珀酰亚胺酯)、BADANTM(6-溴乙酰基-2-二甲基氨基萘)和ACRYLODANTM(6-丙烯酰基-2-二甲基氨基萘)。在其他实施方案中,标记物可以是基于香豆素的染料,例如但不限于酮基香豆素和3,3’-羰基双(7-二乙氨基香豆素)。在其他实施方案中,标记物可以是PyMPO-SETM(1-(3-(琥珀酰亚胺氧羰基)苄基)-4-(5-(4-甲氧基苯基)噁唑-2-基)吡啶鎓溴化物)。
在一些情况下,可以将靶蛋白的一级氨基酸序列中的天然氨基酸残基突变为或置换为另一种能够与二次谐波活性染料结合的氨基酸。本文所述的靶蛋白可包括保守置换。
在其他情况下,二次谐波活性部分(例如,标记物)可以是非天然氨基酸(UAA)。与常规的标记物不同,UAA提供了在埋藏的和暴露的位点处均标记蛋白质的手段。此外,作为蛋白质的固有组分,它们可以相比于连接到氨基酸官能团(例如,半胱氨酸、胺等)上的标记物(如染料)以更高灵敏度和保真度报告结构变化。UAA具有用于使用非线性技术如二次谐波产生来检测蛋白质的超极化性。因此,这些特定的非天然氨基酸也被称为SHAA(“二次谐波氨基酸”)。使用UAA作为探针来检测蛋白质结构构象变化的另一个优点是,该检测可以在体内进行——也就是说,在活细胞中进行。例如,本文所述的方法可以用来检测活细胞中的靶蛋白响应于候选药物的结合所表现出的信号。
任何可超极化的UAA可以作为二次谐波活性部分(例如,标记物)。UAA的实例包括Aladan(Cohen等,2002,Science,296:1700;Abbyad等,2007,J.Phys.Chem.,111:8269,其公开内容通过引用整体并入本文)、丹酰丙氨酸(Summerer等,Proc.Nat.Acad.Sci.U.S.A.,2006,103(26):9785-9789)。在一个实施方案中,所述非天然氨基酸是和频产生活性的(SFG-活性的)。在其他实施方案中,所述UAA不是可超极化的,但具有合适的一个或多个化学官能团,从而使其能够结合二次谐波活性标记染料,例如上述任意染料。在其他实施方案中,所述UAA可包括具有定制的振动性质的探针,用于工程构建到蛋白质内的不连续位点,以通过SFG鉴别位点特异性构象变化。在一些实施方案中,理想的是,用于包含在UAA内的探针部分足够小,以使其不扰乱天然蛋白质结构,并且可以包括但不限于NO、CN、SCN或N3。在一些实施方案中,探针部分提供在约1900至2300cm-1的频谱范围内的独特振动特征(vibrational signature),这与内在蛋白质的振动能够很好地分开。在另一实施方案中,UAA可用于将靶蛋白连接到表面上,使得二次谐波活性部分(例如,标记物)具有相对于表面的净取向。
在一些实施方案中,非天然氨基酸包含同位素探针部分如氘,13C或其他同位素。同位素标记(如氘化)应用的一个说明性实例在Weidner等人,“Sum frequency generation and solid-state NMR study of the structure,orientation,and dynamics of polystyrene-adsorbed peptides,”13288–13293,PNAS,2010年7月27日,vol.107,no.30中阐述,其通过引用全文并入本文。在一些实施方案中,可采用在靶蛋白的一级氨基酸序列中的天然(即,天然存在的)氨基酸或氨基酸残基的同位素标记。在一些实施方案中,天然和非天然氨基酸的同位素标记可一起使用。进而,在一些实施方案中,可以采用任何生化实体(候选物、靶标或两者)的同位素标记。因此,同位素标记不仅可包括氨基酸,还可包括本文提供的生化实体中的任何原子或部分。在一些情况下,同位素标记物或探针还可连接至本文的生化实体上。
相应地,在一些情况下,通过测量工程构建到靶蛋白不同突变体的氨基酸序列内的非天然氨基酸标记物或一系列此类标记物的倾斜角或绝对倾斜角,能够实时地和在真实空间中确定靶蛋白的结构变化。探针可以在靶蛋白内的任何位点处或在其末端,或在其任何结构域中引入。在一些实施方案中,靶蛋白可包括在化学上配备成与UAA共价反应的二次谐波活性标记物。例如,如果引入蛋白质内的UAA是对乙酰基苯丙氨酸(pAcF),则二次谐波活性染料将在其上具有适当的化学性质(chemistry),以便与pAcF共价键合。在另一实施方案中,除了第二UAA以外还引入SHAA,该第二UAA(其通常不是二次谐波活性的)允许蛋白质以定向方式在化学上垂直共价偶联到表面上,该表面用适当的化学物质进行衍生化以便与第二UAA反应。对于高度定向的SH-活性的靶蛋白样品(使用这两种UAA),基线SHG信号和对比度(在构象变化时的信号变化)均可以较之不采用UAA产生SHG信号的靶蛋白更大。
在其他情况下,利用本文公开的任意方法在靶蛋白的氨基酸序列中使用一个或多个UAA使得能够通过确定靶蛋白内一个或多个位点处的一个或多个标记物随时间变化的倾斜角,来确定靶蛋白在与候选别构调节剂结合时经历的实际构象变化。靶蛋白的三维结构可以如下确定:制备蛋白质的一个或多个突变体,其各自含有放置在不同位置上的SHAA探针(即,对于每个突变体中的探针,可以确定每个突变体中探针相对于表面的取向,因此确定侧链的取向,并且可以使用该信息构建整体三维蛋白质结构的模型)。由本领域技术人员已知的空间位阻方法、统计方法、分子动力学、拉马钱德兰图(Ramachandran plot)或能量最低化方法获得的信息可以用于进一步帮助考虑所测量的探针倾斜角来确定该结构。对探针倾斜角的时间分辨测量产生蛋白质的构象变化在实时发生时的运动图像。由于SHG的(以及SFG的)实际上瞬时的响应和灵敏度,只要信号强度足够,特定探针的空间取向(例如,相对于表面的倾斜角或绝对倾斜角)可以在任何感兴趣的时间尺度上实时地测量。
关于在SHG技术中使用UAA的进一步的信息可以在美国专利申请公开第2010/0068144号中找到,其公开内容通过引用整体并入本文。
VIII.界面
在本文所公开的方法的一些方面,靶蛋白与固体表面结合或相对于界面定向,使得与靶蛋白结合的二次谐波活性标记物具有净取向。正是这种净取向在靶蛋白活性位点内结合时或在能够将突变型或野生型靶蛋白的结构稳定为非活性或活性构象的候选物剂结合时能够改变,只要该物剂诱导标记的靶蛋白的结构的构象变化。在一些实施方案中,界面可以由二氧化硅、玻璃、硅、聚苯乙烯、尼龙、塑料、金属、半导体或绝缘体表面,或任何可以吸附或附着生物成分的表面制成。在不同的实施方案中,界面可以是汽-液界面、液-液界面、液-固或固-固界面。在一个实施方案中,该汽-液界面是空气-水界面。在一个实施方案中,该液-液界面是油-水界面。在不同的实施方案中,该液-固界面是水-玻璃界面或苯-SiO2界面。
在一些方面,界面还可以包括生物细胞和脂质体表面。附着或固定可通过多种本领域公知的技术进行。例如,对于蛋白质,表面可使用醛硅烷进行衍生化,以供与生物分子表面上的胺偶联(MacBeath和Schreiber,2000——其相关部分通过引用并入本文)。BSA-NHS(BSA-N-羟基琥珀酰亚胺)表面也可以如下使用:首先使BSA的分子层附着到表面上,然后用N,N’-二琥珀酰亚胺碳酸酯将其活化。BSA上的活化的赖氨酸、天冬氨酸或谷氨酸残基与蛋白质上的表面胺反应。
也可使用支持的磷脂双层,同时使用或不使用膜蛋白或其他膜相关成分,例如,Salafsky等人,Biochemistry,35,14773-14787,1996——其相关部分通过引用并入本文,“Biomembranes”,Gennis,Springer-Verlag,Kalb等人,1992,以及Brian等人,1984,它们的相关部分通过引用并入本文。支持的磷脂双层是本领域公知的,并且有许多技术可用于其制备,其中使用或不使用相关的膜蛋白。这些支持的双层通常必须浸没于水溶液中,以防止其在暴露于空气时破坏。在一些实施方案中,该表面是脂质类似物双层表面。
如果使用固体表面(例如,平面基底、珠子等),其也可以通过多种化学反应进行衍生化,以降低或提高其净表面电荷密度,从而优化对靶蛋白-候选别构调节剂相互作用的检测。在其他实施方案中,固体表面可以是玻璃表面、塑料表面、金属表面、胶乳表面、橡胶表面、陶瓷表面、聚合物表面、聚丙烯表面、聚偏二氟乙烯表面、聚苯乙烯表面或聚乙烯表面(如聚乙二醇表面)。靶蛋白固定于其上的载体可以由广泛的材料构成,例如但不限于生物的、非生物的、有机的、无机的或任何上述这些的组合,以颗粒、链、沉淀物、凝胶、板片、管、球、容器、毛细管、垫、薄片、膜、板或载玻片形式存在。表面可以具有任何适宜的形状,例如但不限于圆盘、正方形、球形或圆形。表面可以优选是平整的,但也可以采取多种替代表面构型。例如,表面可包含凸起的或凹陷的区域,样品(例如蛋白质)位于其上。表面优选地形成刚性支撑,在其上可以形成样品。表面也选择为用于提供适当的吸光特性。例如,表面可以是但不限于聚合的Langmuir Blodgett膜、官能化的玻璃、Si、Ge、GaAs、GaP、SiO2、SiN4、改性硅,或多种凝胶或聚合物中的任何一种,例如(聚)四氟乙烯、(聚)偏二氟乙烯、聚乙二醇、聚苯乙烯、聚碳酸酯或其组合。其他表面材料对于本领域技术人员将是显而易见的。在一个实施方案中,基底是平板玻璃或二氧化硅。
在一些方面,表面可以用公知的技术进行蚀刻,以提供所需的表面特征。例如,通过形成沟槽、V形槽、台面结构等,可以将靶蛋白(如,蛋白质的合成区域)更接近地置于照射光的焦点内。表面也可设置有反射性“镜子”结构以使从中收集的发射最大化。作为另一实例,可以对表面进行蚀刻以形成孔。
在本发明的另一方面,寡聚聚乙二醇(PEG)分子可用于将亲和标记的靶蛋白固定于表面上以供SHG或SFG检测。在一些实施方案中,PEG可以是SAT(PEG4)(N-琥珀酰亚胺基S-乙酰基(硫代四乙二醇)。适于检测SHG信号的PEG化的界面可以通过用寡聚PEG溶液涂覆合适的表面如上述任何表面而制备。在一个实施方案中,表面可以是玻璃。在另一实施方案中,表面可以是氨基封端的硅烷衍生化的玻璃。亲和标签是本领域中常见的,并且可以是,例如,组氨酸标签(如His6标签)、麦芽糖结合蛋白标签、HA标签、生物素标签、硫醇标签或GST标签。在一些实施方案中,亲和标签是具有任意1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20个或更多个组氨酸残基的组氨酸。在一个实施方案中,寡聚PEG分子使用将会与靶蛋白上表达的亲和标签结合的试剂进行修饰。该试剂在组氨酸标签的情况下可以是镍或钴,或者可以是糖(如麦芽糖)、抗体或本领域已知的任何其他能够与亲和标签结合的分子。
IX.试剂盒
本发明提供了一种试剂盒,其包含用于实施本文所述的方法的组合物。例如,试剂盒可包含一个或多个用于偶联至靶生化实体(例如,靶蛋白)的SHG、SFG或DFG-活性部分(如标记物),如任何本文中所描述的SHG、SFG或DFG-活性标记物(如染料标记物)。这些可以包括具有针对特定官能团的不同偶联化学性质的染料标记物,例如但不限于,马来酰亚胺标记物(如,PyMPO-MALEIMIDETM(1-(2-马来酰亚胺基乙基)-4-(5-(4-甲氧基苯基)噁唑-2-基)吡啶鎓甲磺酸盐))、PyMPO-NHS、噁唑标记物、BADANTM(6-溴乙酰基-2-二甲基氨基萘)、PyMPO-SETM(1-(3-(琥珀酰亚胺氧羰基)苄基)-4-(5-(4-甲氧基苯基)噁唑-2-基)吡啶鎓溴化物)以及ACRYLODANTM(6-丙烯酰基-2-二甲基氨基萘)。在其他实施方案中,染料可以是基于香豆素的染料,例如但不限于酮基香豆素和3,3'-羰基双(7-二乙基氨基香豆素)。
在试剂盒中预期还包含供本文公开的任意方法使用的、用SHG、SFG或DFG-活性标记物标记蛋白质的工具和试剂。这些可以包括但不限于,用于测试不同的标记条件的缓冲液组、用于进行标记反应的缓冲液、在其中进行标记反应的管、用于从未结合的标记物中纯化标记的蛋白质的旋转柱和/或凝胶过滤柱,以及用于洗涤标记的蛋白质的缓冲液。
在另一实施方案中,试剂盒可包含一个或多个用于结合靶生化实体(例如,蛋白质)的表面,如本文公开的任何表面。这些表面可以是可重复使用的或消耗性的(即,打算使用一次或有限的次数)。表面可以由玻璃(如载玻片)、塑料、金属、胶乳、橡胶、陶瓷、聚合物(例如但不限于聚丙烯、聚偏二氟乙烯、聚苯乙烯或聚乙烯(例如,聚乙二醇))或任何可以吸附或附着生化实体(例如,生物分子如蛋白质)的表面制成。在一些实施方案中,表面是平滑的。然而,在其他实施方案中,表面可包括预先形成的表面特征,例如但不限于沟槽、V形槽、台面结构和/或孔。此外,表面可以根据任何本文所公开的方法进行预清洗。在另外的实施方案中,当表面不包括预先形成的表面特征时,试剂盒可以包含用于在表面上模板化形成孔的粘合垫圈。
在一个实施方案中,可以针对不同的激发几何形状对包含在试剂盒中的表面进行优化,其中传送或生成非线性光学光的模式基于但不限于TIR(全内反射)、透射或反射中的一种或多种。
在进一步的实施方案中,包含在本文所公开的试剂盒中的表面可以预先涂覆有一种或多种提供适合于连接到靶生化实体(例如,靶蛋白)上的表面化学性质的分子。这些分子可以包括任何本文公开的分子,例如但不限于醛硅烷、BSA-NHS(BSA-N-羟基琥珀酰亚胺)或寡聚聚乙二醇(PEG)。在一个实施方案中,该分子可以包括亲和标签,如任何本文公开的那些标签。例如,表面可以涂覆有镍-寡聚PEG分子以用于将组氨酸-标记的蛋白质固定至表面上。
在另一实施方案中,包含在试剂盒中的表面可以预先涂覆有支持的脂质(例如但不限于磷脂)双层或支持的脂质类似物双层。该试剂盒的该实施方案还包含用于在任何本文所公开的表面上制备支持的脂双层或支持的脂质类似物双层的脂质或脂质混合物。在一些实施方案中,脂质混合物可以包含磷酸胆碱,例如但不限于DOPC(1,2-二油酰基-sn-甘油-3磷酸胆碱)。在一些实施方案中,脂质混合物可以包含但不限于,DDAB(N,N-二硬脂基-N,N-二甲基溴化铵)、DMRIE(N-(1,2-二肉豆蔻基氧基丙-3-基)-N,N-二甲基-N-羟乙基溴化铵)、DODAC(N,N-二油烯基-N,N-二甲基氯化铵)、DOGS(双十七烷基氨基甘氨酰基亚精胺)、DOPE(1,2-sn-二油酰基磷脂酰乙醇胺)、DOSPA(N-(1-(2,3-二油烯基氧基)丙基)-N-(2-(精胺甲酰胺基)乙基)-N,N-二甲基三氟乙酸铵)、DOTAP(N-(1-(2,3-二油烯基氧基)丙基)-N,N,N-三甲基氯化铵)、DOTMA(N-(1-(2,3-二油烯基氧基)丙基)-N,N,N-三甲基氯化铵)中的一种或多种。在进一步的实施方案中,任何脂质混合物的脂质或脂质组分可以包括用于与蛋白质结合的亲和标签(例如但不限于镍化脂质,例如,DOGS-Ni-NTA脂质)。
预期在本文所公开的一种或多种试剂盒中还包含与如下的设备一起使用的信号外围设备,该设备用于检测生化实体之间的相互作用(例如,靶蛋白-候选药物相互作用)和对靶生化实体(例如,蛋白质)构象结构的影响,用于确定靶生化实体(例如,生物分子)与另一种生化实体(例如,候选药物)之间的结合相互作用的特异性。这些可以包括通滤波器、滤色器、干涉滤波器或其他滤谱器(用于波长选择或从高次谐波中分离基本光)、一个或多个偏振光学器件和/或一个或更个用于引导和聚焦光束的反射镜或透镜。以定量测量为目的的机动化旋转台也可包括在本文所公开的试剂盒中。
在另一实施方案中,蛋白质或酶功能所必需的一种或多种辅因子可以包含在本文所公开的试剂盒中。这些可以包括无机辅因子(例如但不限于铜、铁、镁、锰、钼、镍、钴或锌离子)。这些也可包括有机辅因子,例如但不限于焦磷酸硫胺素、NAD+(NADH)、NADP+(NADPH)、磷酸吡哆醛、硫辛酰胺、甲基钴胺素、钴胺素、生物素、辅酶A、四氢叶酸、甲萘醌、抗坏血酸、黄素单核苷酸、黄素腺嘌呤二核苷酸、辅酶F420、三磷酸腺苷(ATP)、二磷酸腺苷(ADP)、三磷酸鸟苷(GTP)、二磷酸鸟苷(GDP)、S-腺苷基甲硫氨酸、辅酶B、辅酶M、辅酶Q、三磷酸胞苷、谷胱甘肽、血红素、核苷酸糖、吡咯并喹啉、醌和/或四氢生物蝶呤。预期在本文公开的任何试剂盒中还包含已知能与一种或多种靶蛋白(例如,泛素或SUMO)结合的一种或多种标记的(如SFG或SHG标记的)肽和/或蛋白质,或一种或多种SFG或SHG标记的抗体或其片段。
在其他实施方案中,试剂盒可包含一种或多种用于向生化实体(例如,蛋白质)中引入SHG-、SFG-或DFG-活性非天然氨基酸(UAA)标记物的组分。这些组分可以包括但不限于无细胞翻译提取物、用于引入特定非天然氨基酸标记物(如,Aladan)的tRNA/tRNA合成酶对和/或反应和洗涤缓冲液。
在另一实施方案中,试剂盒可包含一种或多种用于活细胞标记的组分。这些可以包括但不限于,用于细胞附着的表面、SHG、SFG或DFG-活性标记物、在细胞标记反应中使用的缓冲液、用于除去未反应的标记物的洗出缓冲液以及细胞生长培养基。
在一些实例中,这些组分可允许选择性地在生化实体(例如,靶蛋白)上的一个或多个位置进行标记,例如,在距离蛋白质结构之上或之内的活性位点、结合位点、别构结合位点或任何其他位点(例如,可能经历通过试验筛选的构象变化的任何位点,经历与体内或体外药理学性质相关的构象变化的任何位点,等等)给定距离处(例如,在该位点处或与之相邻)的关键位置进行标记。试剂盒可包括用于在预定位置实现选择性标记的工具。例如,试剂盒可包括用于在同一位置标记多种靶生化实体的工具,这使得能够进行如本文其他地方所述的基于信号特征的筛选和评估。
X.系统
本发明还提供了用于实施本文所述方法的系统。在一些实施方案中,所述系统包含用SHG-、SFG-或DFG-活性部分(例如,在本文所提供的任何组合物、方法或试剂盒中公开的任何SFG-、SHG-或DFG-活性部分,例如,SFG-、SHG-或DFG-活性染料标记物或SFG-、SHG-或DFG-活性非天然氨基酸)标记的靶蛋白。所述系统还包含用于检测靶蛋白-物剂相互作用的设备,其包括:i)基本光的光源;ii)具有表面连接的探针(例如SHG标记的、SFG标记的或DFG标记的靶蛋白)的基底;和iii)用于测量二次谐波或其他非线性光束的强度的检测器。在该系统的一些实施方案中,通过该设备检测物剂与蛋白质上的别构位点的结合,并且可以将检测器所记录的数据记录在固定型或数据存储介质上,该介质可通过用于读取存储介质的系统访问。
在其他实施方案中,该系统还包括单色器(用于波长选择)、通滤波器、滤色器、干涉滤波器或其他滤谱器(用于波长选择或从高次谐波中分离基本光)、一个或多个偏振光学器件、一个或多个用于引导和聚焦光束的反射镜或透镜、计算机控制或软件。该系统可包含具有处理器的计算机,该处理器与存储器可操作地耦合并配置用于执行计算机控制或软件(例如,用以执行体现该软件的算法)。在其他实施方案中,传送或生成非线性光学光(例如,SHG)的模式可以基于以下一种或多种手段:TIR(全内反射)、光纤(连接有或未连接有珠子)、透射(基本光穿过样品)、反射(基本光从样品反射)、扫描成像、共聚焦成像或扫描、用于功率积聚的谐振腔、多通设置。
在所述系统的另一个实施方案中,测得的信息可以采取矢量的形式,其可以包括以下一个或多个参数:光的强度(通常由PMT或光电二极管转换为光电压)、光的波长(由单色器和/或滤波器确定)、时间或位置。所述设备的两种一般配置可以为:图像扫描(例如,基底的成像——作为x,y坐标的函数的强度、波长等)和光谱学(例如,强度、波长等的测量,对一些平整表面或对于细胞、脂质体或其他颗粒的悬浮液)。
可以通过参考以下实施例进一步理解本发明,这些实施例是以举例说明的方式提供的,并不意味着是限制性的。
实施例
实施例1:筛选靶向α-突触核蛋白的候选药物的方法
这里,我们报道了SHG作为配体筛选方法的第一次使用,其采用靶蛋白构象变化的实时读出。我们集中研究了蛋白质α-突触核蛋白(帕金森病(PD)的治疗靶点)的精胺诱导的构象变化。我们探讨了是否可以采用精胺作为工具化合物来鉴别与该蛋白质结合的配体并调节其构象。α-突触核蛋白是140个残基的高度酸性蛋白质,并且是PD的主要靶标。PD是继阿尔茨海默病后第二大常见的神经退行性疾病,并且在美国影响了100-150万人(Lang AE和Lozano AM,New England Journal of Medicine 339(16):1130-1143,1998)。该疾病的两个特征是中脑的黑质中的多巴胺能神经元的损失和神经元中路易体(LB)(主要由α-突触核蛋白的聚集体组成)的存在。α-突触核蛋白的点突变与罕见形式的早发型PD相关,表明该蛋白质牵涉疾病机制(Klockgether T,Cell and Tissue Research 318(1):115-120,2004)。越来越多的证据表明,在患有该疾病的患者的大脑中发现的较大聚集体之前形成的α-突触核蛋白寡聚物在体内可能是神经毒性的(Winner B等人.,Proceedings of the National Academy of Sciences.108(10)):4194-4199,2011)。蛋白质从单体发展为原纤维聚集体的过程是目前严格考察的对象(Norris EH等人,Journal of Biological Chemistry280(22):21212-21219,2005;Karpinar DP等人,EMBO J28(20):3256-3268,2009)。最近的报道表明,该蛋白质在体内采取四聚体结构,意味着分解成单体先于寡聚化和聚集。不论毒性种类如何,单体的α-突触核蛋白是药物的有吸引力的治疗靶标,该药物可使之稳定并防止在最早期的聚集。已对α-突触核蛋白进行了广泛的体外研究(Winner B等人,Proceedings of the National Academy of Sciences.108(10)):4194-4199,2011;Bertoncini CW等人,Proceedings of the National Academy of Sciences of the United States of America102(5):1430-1435,2005;Conway KA,Harper JD,&Lansbury PT,Biochemistry 39(10):2552-2563,2000)。它是天然非结构化的,采取从紧凑到充分伸展的一系列构象(Bertoncini CW等人.,Proceedings of the National Academy of Sciences of the United States of America102(5):1430-1435,2005)。C-末端与N-末端之间的相互作用似乎使蛋白质主要稳定在紧凑的构象。一种假设称当蛋白质暴露于多胺(如精胺)或低pH时,这些相互作用被破坏并且体外聚集的速率大幅增加。另一种针对精胺作用的假设称:结合的精胺使C末端区域的高度负电荷状态降低,从而降低了蛋白质分子之间的静电排斥力并导致聚集速率大幅增加(McClendon S,Rospigliosi CC和Eliezer D,Protein Science18(7):1531-1540,2009)。在任一种情况下,该蛋白质均在聚集前经历构象变化。
为了达到鉴别α-突触核蛋白的结合剂或构象调节剂的目标,我们构建了称为Artemis的基于SHG的筛选仪器,该筛选仪器读取模板化的显微镜载波片。通过该仪器实时检测并报告配体对蛋白质构象的影响。硅酮模板在每个载玻片上限定16个孔;每个孔的测量需要数秒来完成。目前的吞吐量在每天数百次测定的数量级,适合用于片段或所关注的文库的筛选,但我们计划通过实施标准流体学和自动化来将该技术扩大规模至高吞吐量。
我们开发了针对天然非结构化的蛋白质—单体α-突触核蛋白(帕金森病(PD)的靶标)的构象变化的SHG测定。α-突触核蛋白从单体到寡聚物到原纤维的聚集牵涉发病机制,并且被认为之前存在通过与配体结合或pH变化而诱导的单体构象变化。精胺(一种内源性多胺)诱导蛋白质构象变化,该变化使蛋白质的体外聚集速率增加了105倍并且可能在发病机制中起作用。我们采用精胺作为构建α-突触核蛋白的构象变化测定中的工具化合物。我们采用该测定从片段文库中筛选可抑制精胺对蛋白质构象影响的化合物。我们的筛选从文库中鉴别出一种完全消除精胺的影响的化合物。当我们测试其自身对蛋白质构象的影响时,该化合物产生了与精胺相似的构象变化的SHG响应“信号特征”,表明其可能诱导相同的构象并与精胺结合至相同位点。随后我们通过1H、15N杂核单量子相干(HSQR)NMR确认该化合物与单体蛋白质的C末端区域(精胺结合的区域)结合。因为蛋白质一直难以通过传统技术进行筛选,所以已知与α-突触核蛋白结合的化合物较少,因此我们的方法提供了一种鉴别可稳定单体的非聚集构象以及β-淀粉样蛋白(阿尔茨海默病的靶标)的类似构象的化合物的手段。我们发现的化合物是第一个已知与该蛋白的C末端区域结合的化合物,并且可能是治疗先导化合物开发的有用起点。由于SHG原理上可应用于任何靶标,因此这里我们介绍的技术提供了用于鉴别和验证调节蛋白质构象的配体的灵敏且快速的手段,该手段对于基础研究和药物发现具有广阔潜力。
方法
α-突触核蛋白纯化和标记:构建α-突触核蛋白的单位点半胱氨酸突变体(A90C)并用PyMPO-马来酰亚胺(Invitrogen)(我们在先前的研究中使用的SH活性探针)进行标记(Salafsky,Journal of Chemical Physics 125(7),2006;Salafsky,Physical Chemistry Chemical Physics 9(42):5704-5711,2007)。先前已确定突变体(A90C)蛋白质起到类似野生型的作用(Bertoncini CW等人.,Proceedings of the National Academy of Sciences of the United States of America102(5):1430-1435,2005)。根据制造商的说明将标记物偶联至蛋白质并通过凝胶过滤进行纯化。
Artemis SHG筛选仪器:Artemis仪器包含锁模Ti:蓝宝石振荡器,其提供产生二次谐波光(高峰值功率强度)所必需的基本光束。我们采用由Millenia V DPSS激光器(Spectra-Physics Corp.)泵送的Mira 900Ti:蓝宝石超快振荡器(Coherent Inc.)作为基本光源。Ti:蓝宝石振荡器在780nm处产生0.75W的基波功率,其在80MHz下具有约200fs的脉冲持续时间。本实施例中描述的所有实验中的基本光束穿过半波片以选择p-极化,并聚焦到Dove棱镜中以全内反射(TIR)成大小约50μm的斑点。二次谐波光通过聚光透镜进行收集,采用分色镜(dichroic mirror)和滤波器与基本光分离,并导向具有用于光子计数的内置前置放大器(Hamamatsu)的PMT模块。使用定制电子板将信号数字化,并将数据发送到运行控制和数据采集软件(LabView)的计算机。
采用BK7折射率匹配液(Cargille)将载有蛋白质的载玻片耦合至棱镜,并将棱镜自身固定到能够实现10μm随机可寻址精度的1-D平移平台(Renishaw)上。通过经由控制软件使平台平移来依次读取16个孔的每一个。Artemis装配有递送液体团剂的单通道液体注射器(Cavro)。液体团剂体积和递送速度可以变化。在此处描述的实验中,我们采用在1秒内递送10μl体积。测量的参数包括配体注射之前和之后的读取时间、积分时间、平台温度(此处所有实验均保持在室温下)以及平台相对于基本光束线的位置。
α-突触核蛋白测定载玻片制备:使用定制加工的装配夹具将带有粘性背衬的定制硅酮模板(Arrowleaf Research)施于可商购的Xenobind玻璃显微镜载玻片(Xenopore),以在每个载玻片上限定16个直径3mm(体积约15μl)的孔。使限定孔的模板符合标准的96孔板。将PyMPO-马来酰亚胺标记的α-突触核蛋白的储备溶液在磷酸盐缓冲盐水(PBS)中稀释成5μM的最终浓度,并通过在室温下温育30分钟使其吸附到每个孔中的玻璃表面。使蛋白质吸附后,将载玻片放置在Artermis平台上,并通过SHG对每个孔进行扫描以确定每个孔中蛋白质的量。然后,通过用20μL PBS洗涤5次来去除未结合的蛋白质。然后进行洗涤后的孔扫描以确定有多少蛋白质结合至载玻片表面。
Maybridge文库筛查:Maybridge Ro3片段文库含有超过1,000种纯度≥95%以50mM溶解在DMSO中的药效化合物。对于片段筛选实验,将来自文库的单个化合物在PBS中稀释并以1mM(4%DMSO)的最终浓度置于每个孔中。为确保不发生缓冲液错配,在筛选中使用的洗涤缓冲液为PBS+4%DMSO。然后将载玻片装载到仪器上,实时测量基线信号,并在读取SHG信号的同时注射5mM的精胺。在这些实验中,监测基线信号8秒,随后注射精胺并另外记录SHG信号8秒。
剂量-响应曲线拟合:为确定精胺(Sigma)、亚精胺(Sigma)以及化合物A的剂量-响应曲线,使用每种化合物的系列稀释液来制备储备溶液。将每种化合物添加至孔中以得到指定的最终浓度。对于精胺,所测试的浓度范围为10μM、200μM、400μM、600μM、800μm、1mM和5mM。对于亚精胺,浓度为10μM、62.5μM、125μM、250μM、500μM、1mM和30mM。对于化合物A,浓度范围为5μM、32.5μM、62.5μM、125μM、250μM、500μM和1mM。然后将浓度转化为对数尺度,并在GraphPad Prism V5中采用log(激动剂)与响应的具有可变斜率的非线性回归拟合将数据作图。将数据归一化,使得最低浓度和最高浓度的值分别等于0和100%。通过软件计算EC50值。
竞争测定:对于精胺-亚精胺竞争测定,将最终浓度1mM的精胺置于含有5μM标记的α-突触核蛋白的每个孔中,并测量SHG强度的变化。然后将蛋白质在1mM精胺的存在下在室温下温育25分钟。25分钟后,向每个孔添加3mM亚精胺并测量产生的SHG强度的变化。类似物-精胺竞争测定如上述进行,不同之处在于首先将每种单独的类似物与α-突触核蛋白一起温育25分钟并随后向孔中添加1mM精胺。
对于精胺-化合物A竞争测定,将5mM精胺置于含有5μM标记的α-突触核蛋白的每个孔中,并测量SHG强度的变化。使蛋白质在精胺的存在下平衡一分钟,并向每个孔添加100μM或1mM化合物A并测量SHG强度的变化。
定量SHG强度的变化:对每种化合物进行对照实验,以凭经验确定每种化合物产生最大响应(Tmax)所需的时间范围。对于除使用四磺酸酞菁(PcT;Sigma)的那些实验之外的所有实验,如通过SHG测量的最大响应均在添加化合物后的30s内发生。对于PcT实验,最大响应发生在添加化合物后约45s。
为计算变化百分比,根据以下等式,从Tmax时的SHG强度中减去即将注射前(T0)测量的SHG强度并然后除以T0:SHG%=(ISHG@Tmax-ISHG@T0)/ISHG@T0。
对照缓冲液注射用于确定SHG强度变化的阈值,并以与上文相似的方式进行计算。然后通过从由化合物添加而产生的SHG强度值中减去缓冲液阈值来报告每种化合物的净SHG强度变化。所得值被报告为由于化合物添加而引起的SHG强度的变化百分比。
结果
精胺诱导α-突触核蛋白的构象变化:为了通过二次谐波产生(SHG)筛选抑制α-突触核蛋白构象变化的新型化合物,我们开发了一种采用精胺的测定。精胺是一种涉及许多细胞过程的多胺,且先前已被证明诱导导致体外聚集速率增加的α-突触核蛋白构象变化。在我们的测定中,定制组装的显微镜载玻片的每个孔均载有5μMα-突触核蛋白,该浓度低于寡聚物形成的阈值(Fernandez CO等人.,EMBO J23(10):2039-2046,2004),并且实时测量精胺添加前后的SHG信号。添加5mM精胺时,SHG信号强度立即增加(图1A)。我们测量得到SHG强度变化为36.26±4.97%。如通过NMR证明的,该变化对应于由α-突触核蛋白的C-末端与精胺结合而诱导的已知构象变化(Bertoncini CW等人,Proceedings of the National Academy of Sciences of the United States of America 102(5):1430-1435,2005)。通过使用SHG,我们获得了精胺与α-突触核蛋白结合的剂量-响应曲线(图1B),并确定精胺的EC50为0.84mM(logEC50-3.07)[重复值:0.79mM(logEC50-3.13)]。
亚精胺(一种与精胺紧密相关的多胺)也已知结合α-突触核蛋白并诱导构象变化。作为另外的对照,我们测试了其对蛋白质的影响,并发现它也诱导α-突触核蛋白的构象变化,且其SHG信号特征与精胺的信号特征相似(图1C)。3mM剂量的亚精胺导致SHG强度19.88±2.37%的增加。接下来我们确定了亚精胺的剂量-响应曲线(图1D)。通过SHG,我们测得亚精胺的结合亲和性为1.11mM(logEC50=-2.95±0.19)。为了验证我们的SHG衍生的剂量响应曲线的EC50结果,我们采用了精胺与亚精胺之间的竞争测定。在竞争测定(图1E)中,向含有α-突触核蛋白的孔添加1mM精胺并测得SHG强度增加25.58±4.33%(重复值:30.23±3.69%)。将α-突触核蛋白在1mM精胺存在下温育25分钟后,向样品中添加3mM亚精胺并记录SHG强度5.1±0.81%的变化。鉴于添加3mM亚精胺单独诱导SHG强度约20%的增加,在精胺存在下观察到的相对小(约5%)的增加表明精胺比亚精胺以更高的亲和性与α-突触核蛋白结合,这与我们通过实验得到的剂量-响应曲线一致。
二次谐波产生作为分子筛选的平台:已经证明我们观察到的SHG信号变化与精胺或亚精胺诱导的构象变化相对应,我们试图筛选将抑制精胺诱导的α-突触核蛋白构象变化的新型化合物。通过采用基于SHG的竞争测定,我们针对其在添加精胺时抑制α-突触核蛋白的构象变化的能力,一式两份对来自Maybridge Rule of Three(Ro3)片段文库的超过1000种配体进行了筛选。每个孔载有5μMα-突触核蛋白,并在测定缓冲液中与1mM来自Maybridge Ro3文库的一种片段化合物一起预温育30分钟。然后向每个孔中添加5mM精胺,并测量所产生的SHG信号变化。如通过SHG测量的,1,000种化合物的整个文库中仅一种化合物—化合物A,在1mM的浓度下完全阻止了精胺诱导的构象变化(-1.39±1.05;图2A)。有趣的是,图2B所示的化合物A与精胺在电荷和长度上有相似之处。如图2C所示(右侧的迹线),向α-突触核蛋白添加1mM化合物A立即改变了SHG基线信号。我们测得SHG强度变化为31.67±4.31%,并如与精胺的响应相似的,采用方向(相对于基线)、大小和速率将它的SHG响应分类(图3D)。
接下来,如通过SHG测量的,我们确定了化合物A是否能够诱导α-突触核蛋白的构象变化。如图3A所示,向α-突触核蛋白添加1mM化合物A立即改变了SHG基线信号。我们测得SHG强度变化为30±2%(重复值:31.67±4.31%),其方向(相对于基线)、大小和动力学与精胺的响应相似。该结果表明:化合物A诱导的α-突触核蛋白构象变化与精胺诱导的α-突触核蛋白构象变化相似。接下来我们对化合物A进行滴定并通过SHG测量其剂量-响应(图3B)。由此我们确定EC50为97.8μM(logEC50-4.01±0.28)。因其Kd为约100μM,化合物A具有比精胺高至约5.5到8倍的α-突触核蛋白亲和性。
鉴于化合物A具有比精胺高得多的α-突触核蛋白亲和性,化合物A在竞争与α-突触核蛋白结合方面应胜过精胺。为了确定化合物A是否可以在竞争与α-突触核蛋白结合方面胜过精胺,我们进行了竞争测定,在该测定中将5mM精胺添加至含有α-突触核蛋白的孔中并使其平衡1分钟。单独的精胺添加导致SHG强度37.44±6.40%的增加(图3C)。将α-突触核蛋白在5mM精胺存在下温育后,向每个孔中添加1mM或100μM化合物A并记录SHG强度的变化。向与精胺预结合的α-突触核蛋白添加1mM化合物A导致SHG强度40.69±5.81%的增加。这与100μM化合物A的添加形成对比,其导致SHG强度3.18±1.50%的增加。这些数据表明,化合物A由于对其靶标更高的亲和性而在竞争与α-突触核蛋白结合方面胜过精胺。进一步的比较在图3D中示出。
来自我们的SHG实验的结果表明,化合物A与α-突触核蛋白结合并诱导与精胺结合相似的构象变化,但其对α-突触核蛋白具有更高的亲和性。接下来,我们想要确定化合物A是否与精胺结合至α-突触核蛋白上的相同位点。为此,我们转而使用独立结构技术2-D HSQR NMR来确定与α-突触核蛋白复合的化合物A的结构。
我们的SHG与NMR数据的结合表明,化合物A在与精胺相同的位点处与α-突触核蛋白结合(图6A)但具有更高的亲和性,由此验证了我们初始筛选的结果,并将SHG确认为结构筛选技术。
通过NMR波谱法分析了α-突触核蛋白与化合物A之间的相互作用。NMR共振是蛋白质-蛋白质和蛋白质-配体相互作用的高度敏感的探测,并因此可以提供相互作用界面和结合亲和性的详细描述。将化合物A添加至15N标记的α-突触核蛋白中,并且化学位移的变化遵循α-突触核蛋白的二维1H–15N相互关系光谱(图6B)。观察到α-突触核蛋白的残基G111到A140的NMR信号的显著变化(图6B和图6C)。在精胺添加至α-突触核蛋白时已经观察到非常相似的扰动图谱,表明化合物A和精胺均与α-突触核蛋白的C末端结构域结合。
采用顺磁弛豫增强(PRE)对化合物A结合至溶液中由α-突触核蛋白占据的全体构象上的结果进行了研究。特异性附着的顺磁性氮氧自由基与附近的质子(小于约)之间的相互作用导致其NMR信号由于横向弛豫率的增加而变宽。这种影响对电子-质子距离具有r-6的依赖性,并因此使得能够检测蛋白质中的长程相互作用。为了观察在化合物A存在下α-突触核蛋白内的瞬时长程接触,我们在α-突触核蛋白的位置90处附着了顺磁性MTSL标记物。化合物A不存在和存在时MTSL-A90Cα-突触核蛋白的PRE谱的比较揭示了在残基40-46和C末端区域覆盖残基124-136处减少的顺磁性变宽(图6D)。因此,化合物A的结合使α-突触核蛋白中的瞬时长程相互作用减弱。结合精胺诱导的α-突触核蛋白聚集的加速,在与精胺结合时α-突触核蛋白中长程相互作用的减弱也已报道。因此,进一步测试了化合物A对α-突触核蛋白聚集的增强。与通过SHG和NMR鉴别的相似结合模式一致,化合物A也显著缩短了α-突触核蛋白原纤维化的延滞期(图7A)。存在(图7B,左侧)和不存在(图7B,右侧)化合物A时获得的α-突触核蛋白原纤维的形态相似。因此,我们的SHG和NMR数据的结合表明:化合物A与α-突触核蛋白C末端结合。此外,NMR和PRE数据表明,化合物A结合至与精胺相同的位点,或结合至与精胺的位点有显著重叠的位点,并且通过以与针对精胺观察到的相似的方式减弱长程相互作用来影响蛋白质的构象,由此确认了初始筛选的结果并确认SHG为结构筛选技术。
化合物A是精胺诱导α-突触核蛋白的构象变化能力的选择性抑制剂:我们在1mM的片段浓度下,使用精胺响应消除的命中标准(例如,添加后在基线的1%以内),采用Maybridge Ro3文库所进行的筛选中仅鉴别出一种化合物。为了探索在不太严格的条件下来自我们文库的化合物A的化学类似物的效果,我们采用程序Acelot SimFinder(acelot.com/SimFinder.html)来排出前五种化合物。如图4A所示,这些类似物在结构上和化学上与化合物A相似。我们首先确定了这些类似物的任一种在暴露于α-突触核蛋白时,其自身是否会诱导SHG强度的变化。每种单独的类似物以1mM添加均导致SHG强度的增加(图4B;表1)。尽管SHG强度的增加表明α-突触核蛋白的构象变化,但SHG信号变化的大小显著低于针对精胺和化合物A所报道的值(t-检验)。由于这些变化的SHG响应相对于基线在幅度和方向上是相似的,因此较低的大小可表明导致相同构象变化(而不是与化合物A诱导的构象变化不同的构象变化)的较低的潜能。例如,α-突触核蛋白靶标可以以相同的方式进行标记,但由不同结合实体诱导的构象变化可发生在每个α-突触核蛋白靶标之上或之内的不同位置(并因此在距离标记物不同的距离处)。α-突触核蛋白的构象状态的变化可能与已知的药理学性质或已知的功能效果相互关联。在一些情况下,该相互关系对于α-突触核蛋白上的给定位置上的构象变化可能具有特异性。因此,远离标记物位置发生的构象变化可能不与测定的已知药理学性质或已知功能效果相关。
与图4B的相似比较在图4D中示出,其中化合物A与5种类似物一起示出。
为了更好地理解化合物A类似物数据的结果,我们通过NMR测试了类似物1、2和5与α-突触核蛋白的结合。对如在图4E中看到的化学位移的差异的分析表明,与化合物A相比,类似物1和2与α-突触核蛋白的结合更弱。
表1:化合物A类似物数据的定量
接下来,我们进行了竞争测定,在该测定中将每一种类似物在α-突触核蛋白的存在下温育25分钟。25分钟后,向每个孔中添加1mM精胺并记录SHG强度的变化。对于所有类似物,向在类似物存在下温育的α-突触核蛋白中添加精胺导致SHG强度增加(图4C;表1)。这些数据表明,尽管类似物在化学上和结构上与化合物A相似,但其诱导α-突触核蛋白聚集的能力较低并且相比于精胺和化合物A其对α-突触核蛋白的亲和性较低。此外,来自我们的类似物竞争测定的结果确认了化合物A是Maybridge Ro3文库中唯一的竞争性突触核蛋白聚集剂,从而确认了我们的初始筛选。
四磺酸酞菁产生不同的SHG信号特征:我们的化合物A的数据表明其与精胺结合在α-突触核蛋白上的相同位点上,产生已知增加α-突触核蛋白体外聚集速率的构象。因为α-突触核蛋白聚集的增加与神经退行性疾病的发病机制有关,所以我们想确定与精胺相比已知的α-突触核蛋白聚集抑制剂—四磺酸酞菁(PcT)是否产生不同的SHG信号特征。如图5A所示,我们观察到当用10μM PcT处理时,α-突触核蛋白的SHG强度降低83.74±6.41%(重复值:-79.6±2.7%)。此外,相比于诱导α-突触核蛋白的聚集倾向构象的其他分子,PcT处理的SHG强度迹线(图5A)产生了完全不同的SHG信号特征。如图5B所示,当用10μM PcT处理时,测得α-突触核蛋白的SHG强度的变化为-79.6±2.7%。
这些数据表明,可以通过选择产生与PcT相似的SHG信号特征的化合物来鉴别新型α-突触核蛋白聚集抑制剂。
最后,在硫黄素T测定中对化合物A进行测试(图7A),并发现化合物A将α-突触核蛋白体外聚集速率增加了约两倍,这与精胺存在时所观察到的增加相同。因此,该化合物具有与精胺几乎相同的功能效果。如上文所讨论的,该化合物也诱导与精胺相同的构象变化。
讨论
药物发现的关键限制是有效搜索化学空间以鉴别诱导临床上重要靶蛋白的构象变化的配体的能力。在此处所描述的研究中,我们呈现了使用二次谐波产生作为用于鉴别构象调节剂的结构技术的第一个实施例。我们筛查了含有超过1,000种化合物的片段文库,并鉴别出抑制精胺诱导的α-突触核蛋白构象变化的单个化合物。该化合物—化合物A,被证明其自身引起构象变化并且与精胺相比以更高的亲和性与α-突触核蛋白结合,表明该配体引起了与精胺相同的构象变化。随后我们采用NMR证明化合物A在与精胺相同的位点与α-突触核蛋白结合并诱导相似的构象变化。
二次谐波产生作为药物发现的新平台:该研究表明SHG是一种用于确定配体作用机制的有力方法,特别是当已知的构象调节剂(如精胺)可用于比较目的时。有可能通过我们的基于竞争的筛选发现根据阻止精胺诱导的α-突触核蛋白构象变化的作用机制分类的两类配体。第一类配体为在精胺结合位点处或其附近结合的位阻抑制剂。第二类配体为诱导α-突触核蛋白构象变化且阻止精胺结合的变构抑制剂。在该研究中呈现的NMR数据确认化合物A是第一类抑制剂中的成员。
然而更令人惊讶的是,我们从Maybridge Ro3文库中仅鉴别出一种化合物。尽管精胺和化合物A的长度和电荷相似,但该文库中的多个化合物也与化合物A具有结构相似性。这些微小的结构差异包括化合物A中的5元环转化为6元环以及其他侧链的存在。在我们的测定中,SHG将化合物A鉴别为α-突触核蛋白的精胺诱导构象变化的特异且有效的抑制剂以及将其与紧密相关的分子相区分的能力证明了该技术的灵敏性,并且证明了SHG作为筛选平台的有效性。
SHG信号特征作为分子筛选的工具:四磺酸酞菁(PcT)是一种已知的α-突触核蛋白体外聚集抑制剂。尽管PcT具有抗聚集的作用,但其并不是用于治疗神经退行性疾病的临床相关化合物。然而,在我们的测定中,它具有用于鉴别抑制α-突触核蛋白聚集的新型化合物的潜力。这是因为每种分子均产生关于相对于基线的信号变化方向、变化大小和动力学的“SHG信号特征”。这些信号特征提供了一种比单独的结合信息更加严格的将配体分类的手段。此外,通过将分子的信号特征与由已知调节剂产生的信号特征进行比较,我们可以对该分子的作用机制做出推定分类(在相同的标记和约束条件下)。我们还可通过SHG信号特征筛选化合物文库,以尝试鉴别产生与已知构象调节剂相似的信号特征的化合物。例如,该方法可用于针对诱导与PcT相似的α-突触核蛋白构象变化的潜在抗聚集化合物来筛查Maybridge Ro3文库。
实施例2:靶向α-突触核蛋白的候选药物的差异筛选
化合物A及其类似物(图4A)组成具有6种结构上(和化学上)相似的候选生化实体的文库。可使用表1中的化合物A及其类似物的仅含化合物(%位移或%变化)SHG响应来进行差异筛选。当暴露于α-突触核蛋白时,化合物A及每种类似物均诱导SHG强度的变化。每种候选生化实体产生不同的SHG信号特征。1mM化合物A的添加导致SHG强度的增加(表1)。1mM每种单独的类似物的添加也导致SHG强度的增加(图4B;表1)。表1中的信号特征响应通过如下方法产生:测量α-突触核蛋白的第一构象状态由此产生SHG基线,使6种结构相似的候选生化实体中的每一种与α-突触核蛋白接触,以及测量α-突触核蛋白的第二构象状态,由此产生6种候选生化实体的SHG信号特征。这些SHG信号特征分别指示α-突触核蛋白的构象状态变化(全部6种信号均显示SHG强度增加)。而且,每种候选生化实体产生不同的α-突触核蛋白(靶生化实体)构象状态变化。
可将表1中的SHG信号特征彼此相互比较(例如,为确定相似性)。例如,可采用计算机可执行程序分析SHG信号特征。在一些情况下,可以比较一个时间序列中的连续SHG信号特征,并将下一个SHG信号特征与根据先前步骤中的比较选择的生化实体进行比较。在一些情况下,可将任意两种或更多种SHG信号特征进行比较。在一些情况下,可将文库中全部6种生化实体的SHG信号特征彼此相互比较。在一些情况下,可将SHG信号特征与具有最高、最低或其他限定参考值的SHG信号特征进行比较。本文所述的差异筛选可在例如,不存在已知调节剂(例如,被批准的药物)的情况下使用(例如,针对一些固有无序蛋白质)。在一些实例中,可进行连续或同时比较以在候选生化实体中选择生化实体。
在该实施例中,通过测量SHG信号的大小相对于SHG基线的变化(例如,靶生化实体构象状态的终点变化;或者,可测量靶生化实体构象状态的动力学(实时)变化),以及SHG信号的方向相对于SHG基线的变化(SHG参数)来产生SHG信号特征。在一些情况下,尽管各不相同,但通过采用一个或多个SHG相似性参数,5种类似物的至少一个亚组的SHG信号特征可被分类(成对地或全部)为彼此相似的。例如,5种类似物的SHG信号的大小相对于SHG基线的变化的差异彼此在50%以内,且文库中全部6种生化实体的SHG信号的方向相对于SHG基线的变化的差异相同。
实施例3:靶向α-突触核蛋白的获批准药物的SHG检测
在本实例中,α-突触核蛋白在暴露于多巴胺(其前药左旋多巴(获批准的药物)的代谢物)时引起信号变化。左旋多巴(也称为L-多巴,在大脑中转化为多巴胺)仍是用于治疗帕金森病的金标准。α-突触核蛋白的异常折叠和聚集被认为是该疾病的主要病因。左旋多巴是帕金森病相关症状的最有效治疗剂。然而,左旋多巴是一种前药(即,药物的前体化学化合物)并且是多巴胺(该药物的生物活性形式)的前体。
实验条件
由于天然的α-突触核蛋白缺乏半胱氨酸残基,因此将丙氨酸到半胱氨酸的突变(A90C)引入到α-突触核蛋白中以产生用于染料结合的位点特异性肽。然后,采用标准技术用PyMPO-马来酰亚胺标记A90Cα-突触核蛋白,并且被修饰的肽的位置通过质谱法被验证为是半胱氨酸90。
对于用α-突触核蛋白进行的SHG实验,将PyMPO-马来酰亚胺标记的A90Cα-突触核蛋白的储备溶液在50mM Hepes,pH 7.4、100mM NaCl中稀释成5μM的最终浓度。然后,通过在室温下温育一小时,将5μM蛋白质溶液固定到醛衍生的载玻片上。在50mM Hepes,pH 7.4、100mM NaCl中进行对照(缓冲液)和化合物注射。
实验通过包含锁模Ti:蓝宝石振荡器的仪器进行,该振荡器提供产生二次谐波信号(高峰值功率强度)所必需的基本光束。我们采用由Millenia V DPSS激光器(Spectra-Physics Corp.)泵送的Mira 900Ti:蓝宝石超快振荡器(Coherent Inc.)。Ti:蓝宝石振荡器在780nm处产生约0.7W的基波功率,其在80MHz下具有约150fs的脉冲持续时间。基本光束穿过半波片以选择p-极化(本实施例中所有实验均采用),并聚焦到Dove棱镜中以全内反射(TIR)成大小约50μm的斑点。二次谐波光通过聚光透镜进行收集,采用分色镜和滤波器与基本光分离,并导向具有用于光子计数的内置前置放大器(Hamamatsu)的PMT模块。使用定制电子板将信号数字化,并将数据发送到运行控制和数据采集软件(LabView)的计算机。
结果
图8和图9示出了多巴胺处理对α-突触核蛋白构象的影响。
图8示出了添加多巴胺时α-突触核蛋白SHG强度的动力学(实时)变化的实例。1mM多巴胺的添加(图8中的箭头所指示的)导致α-突触核蛋白的(归一化的)SHG强度相对于注射前的水平(在由箭头标记的注射之前的时间)增加。SHG强度的变化是多巴胺结合时α-突触核蛋白构象变化的结果。
图9是单独添加缓冲液(左侧)或添加1mM多巴胺(右侧)时α-突触核蛋白的SHG强度的平均变化的定量。对照(缓冲液)注射时α-突触核蛋白的SHG强度的平均变化百分比为-1.74±0.57%。误差条=SEM,N=4。注射1mM多巴胺时α-突触核蛋白的SHG强度的平均变化百分比为3.375±1.13%。采用非配对t检验将P值(图9中**指示的)确定为统计学显著的(P值=0.0067)。
讨论
图8和图9中的SHG结果(例如,动力学或终点SHG信号特征、信号或信号差异)可用于通过例如选择产生与多巴胺相似的SHG信号特征的化合物来鉴别新型候选药物(例如,通过将多巴胺的SHG结果与候选药物的SHG结果进行比较),以替代或改善获批准药物的活性形式。与批准药物的活性形式相比,这些备选药物可表现出相似的或改善的体内和体外药理学性质、表型和/或作用机制。例如,如果候选药物表现出与多巴胺相同的SHG响应,则它可能表现出基本相似的治疗效力和/或药理学(或药代动力学)性质。在一些情况下,此类新型候选药物可表现出改善的毒性和/或临床安全性质。左旋多巴的体内和体外药理学性质,诸如药代动力学谱、表型变化、效力和其他临床数据等的说明性实例在David J Brooks,“Optimizing levodopa therapy for Parkinson’s disease with levodopa/carbidopa/entacapone:implications from a clinical and patient perspective,”Neuropsychiatr Dis Treat.2008年2月;4(1):39–47和其中的参考文献中列出,这些文献通过引用全文并入本文。
实施例4:结构-活性关系(SAR)/编目的确定
采用包含Raf激酶的超过200个拷贝的表面阵列,对包含200种不同的化合物和索拉非尼(每一种均具有400到500g/mol的分子量、三个芳香族基以及2-5个氮原子)的文库的结构-活性关系进行评估。通过本申请中所描述的单次谐波产生(SHG)方法来测量Raf激酶的构象状态,以在不存在任何化合物的情况下建立基线。然后,使文库中的每种化合物与在阵列上的Raf激酶的单个拷贝接触。通过SHG方法测量单个激酶靶标的每个拷贝的构象状态。根据与Raf激酶接触时构象状态的变化确定每种化合物的SHG信号特征。索拉非尼具有包含在正方向上30%变化(即,大小)的SHG信号特征。将这些化合物根据其SHG信号特征和共同的结构参数分组,以产生结构-活性关系(SAR)目录。例如,将信号特征的大小与索拉非尼信号特征相差10%以内的8种化合物编目为“400到500g/mol的分子量、三个芳香族基团、2-5个氮原子以及+27%到+33%大小的信号特征”。根据该SAR谱,进一步测试了这些化合物对Raf激酶的抑制活性,并且发现其中的三种化合物在纳摩尔浓度下确实表现出抗-Raf活性。这样,SHG信号特征可提供测定化合物性质的额外的手段,其中可采用SHG信号特征根据结构-活性关系将化合物进行分类。
尽管本文中已经示出并描述了本发明的优选实施方案,但对于本领域技术人员显而易见的是这些实施方案仅以示例的方式提供。本领域技术人员在不脱离本发明的情况下现将想到多种变化、改变和替代。应当理解本文中所述的本发明实施方案的各种替代方案可用于实施本发明。目的在于以下述权利要求限定本发明的范围,并由此涵盖这些权利要求范围内的方法和结构及其等同物。
- 还没有人留言评论。精彩留言会获得点赞!
- 靶向B细胞受体复合物膜结合IgM的抗体及其用途的制作方法与工艺
- 一种肿瘤靶向细胞药物载体及其应用的制作方法与工艺
- 具有肿瘤细胞靶向性的超细磁性核壳纳米材料及其制备和应用的制作方法与工艺
- 具有肿瘤细胞靶向性的超细磁性核壳纳米材料及其制备和应用的制作方法与工艺
- 一种检测癌细胞内铜离子的癌细胞靶向荧光探针的制作方法与工艺
- 一种利用细胞通透性差异治疗皮肤癌的靶向药物的制作方法与工艺
- 一种基于细胞源性微囊泡的肿瘤靶向递送载体及制备方法和应用与流程
- 靶向CLDN6的免疫效应细胞及其制备方法和应用与流程
- 一种检测靶向CD33的CART细胞中CAR表达的荧光定量试剂盒的制作方法与工艺
- 靶向HBV核心启动子的抗HBV药物筛选细胞模型及其构建和应用的制作方法与工艺