一种石榴皮止泻散的定性与定量检测方法与流程
本发明涉及一种石榴皮止泻散的定性与定量检测方法。
背景技术:
石榴皮具涩肠止泻,止血,驱虫的功效。是一种常用中药材。石榴皮止泻散是由石榴皮药材粉碎成中粉,直接分装成不同规格的袋装即可,为动物保健药,用于猪腹泻,黄白痢。一天用量是3~15g。
石榴皮止泻散的组方与制备工艺如下:
【处方】石榴皮1000g
【制法】取石榴皮,粉碎,过筛,混匀,制成散剂1000g,即得。
石榴皮主含鞣质,多以水解鞣质为主,缩合鞣质类较少。水解鞣质又分为没食子鞣质和鞣花鞣质,没食子鞣质是没食子酸与糖类形成的酯类衍生物;鞣花鞣质是鞣花酸与糖类形成的酯类衍生物,同时,没食子鞣质和鞣花鞣质均可进一步酯化或者通过氧化交联形成更复杂的水解鞣质。所以中国药典一部从2005年版开始,一直采用分光光度法,测定鞣质含量,2015年版才增加上鞣花酸的HPLC含量测定。
其鞣质的具体测定方法是:以没食子酸为指标,通过与磷钼钨酸反应生成深蓝色,在在760nm测定其总酚含量,以总酚含量减去不被干酪素吸附的非鞣质的多酚量,即为鞣质含量。操作步骤是:首先要制备没食子酸对照品溶液,接着制备标准曲线,第三步要制备供试品溶液,该溶液必须放置过夜,鞣质才能提取完全,第四步比色测定供试品溶液中的总酚含量,第五步保温制备不被干酪素吸附的非鞣质多酚样品,并测定其含量。总酚含量减去不被干酪素吸附的多酚含量,即为鞣质含量。实际上测定的是:以对照品没食子酸反应物颜色为参照的,用样品颜色的吸收度计算的含量,到底样品中所含的鞣质与没食子酸是什么比例关系,无法考证。
中国药典2015年版一部鞣花酸的含量测定方法如下:
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.2%磷酸溶液(21∶79)为流动相;检测波长为254nm。理论板数按鞣花酸峰计算应不低于5000。
对照品溶液的制备 取鞣花酸对照品适量,加甲醇制成每1ml含20μg的溶液,即得。
供试品溶液的制备 取本品粉末(过3号筛)0.2g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理(功率150W,频率40kHz)40分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法 分别精密吸取对照品溶液5μl、10μl,供试品溶液各5~10μl,注入液相色谱仪,测定,用外标两点法计算含量,即得。
曾按药典鞣花酸的方法测定石榴皮止泻散的含量,出现鞣花酸波峰拖尾,峰型不对称,且样品浓度高,波峰面积大,流动相pH值在2以下,属于易损色谱柱、非要求范围,无法直接采用,需对方法进行改进。
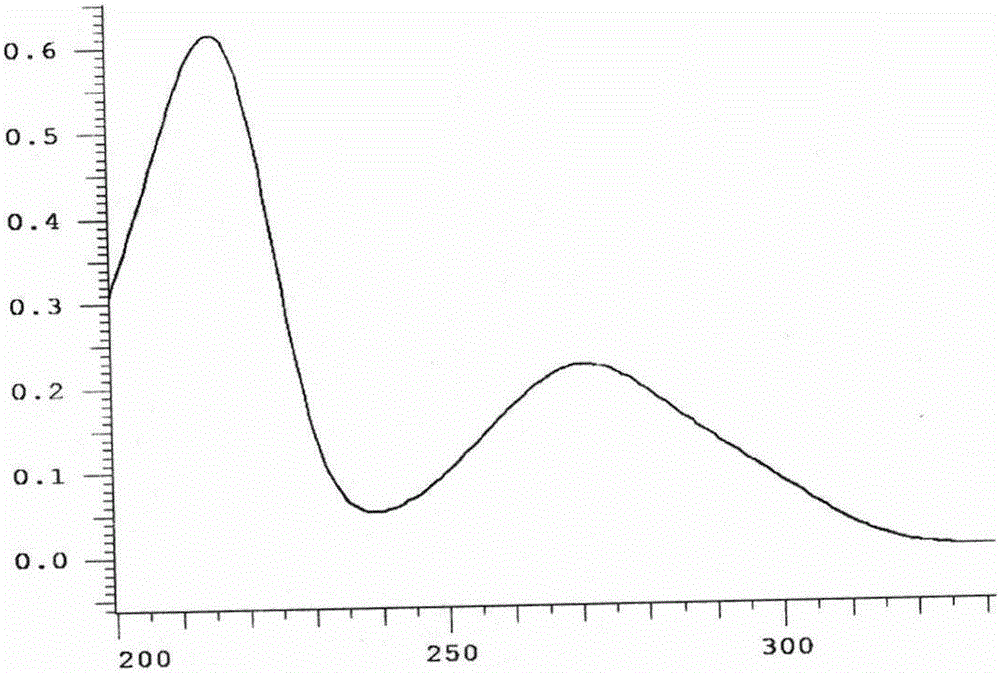
查阅文献得知,有采用高效液相法测定石榴皮中没食子酸含量的报道,但多数都是测定的石榴皮中游离没食子酸,含量低微约1mg/g【1】;检测波长是270nm、273nm或280nm,其波峰高度仅是215nm的1/3(见图2),且流动相多采用缓冲盐【2】,即便不是缓冲盐的,有机相的比例为3%、流速为0.8ml/min,没达到药典限定的5%和1ml/min范围。这样就导致样品取样量大,干扰成分多,缓冲盐或较低的有机相都会降低仪器与色谱柱的寿命。也有测定总没食子酸的报道【3】,但样品前处理不科学,即取石榴皮粉末20g,加盐酸(1→9)250ml,乙醚250ml,回流水解2小时,过滤,提取2次,合并滤液蒸干,甲醇定容至100ml,取续滤液,作为供试品溶液。相当每1ml供试品溶液中含石榴皮药材0.2g。从方法的文字,非专业技术人员很难判断合并的滤液是盐酸水部分,还是乙醚部分。测定的13个产地不同石榴皮中总没食子酸含量在1.30~4.64mg/g之间,平均为2.92mg/g。
我们就是在上述背景条件下,为以简便、快捷、准确、高标准控制石榴皮止泻散的质量,对其进行了质量标准的探讨研究,发明了石榴皮止泻散的定性与定量检测方法。
技术实现要素:
(1)本发明采用薄层色谱法,以石榴皮对照药材为对照,鉴别了其散剂中相对应的特征点,方法简便,只需2ml60%甲醇超声对照药材和供试品,上清液点样,展开,紫外光灯254nm下检视,即可(图1);
(2)采用高效液相色谱法,以体积比为3∶5.4∶91.6的乙腈-甲醇-0.1%磷酸为流动相,在215±1nm处(图2),测定了六批石榴皮止泻散中总没食子酸,含量在3.76mg/g~13.90mg/g之间,平均值为9.08mg/g,波峰分离良好,在三种不同的色谱柱上,峰保留时间都在10分钟左右,进样一针,运行15分钟,没食子酸对照品进样量0.05μg,其波峰面积可达50万,简便、快捷、准确,灵敏度是报道方法的3倍以上。
(3)采用高效液相色谱法,以体积比为18∶5∶77的乙腈-甲醇-0.1%磷酸为流动相,在254nm±1nm处(图10),测定了样品中鞣花酸含量,流动相由体积比21∶79的乙腈-0.2%磷酸溶液修订为体积比18∶5∶77的乙腈-甲醇-0.1%磷酸后,提高了波峰对称性,使流动相的pH值提升到2以上,减少了对色谱柱的损坏;提取溶剂由甲醇修订为80%甲醇,鞣花酸含量提高9.9%,测定数值更趋于样品的真实含量(见表8),方法的准确性提升;鞣花酸的进样量调整为0.04μg,波峰面积在50力,既保证数值准确性,又延长 了色谱柱寿命。
通过方法学考察,没食子酸的进样量在0.012298~0.196773μg,与峰面积呈良好的线性关系,回归方程为:Y=10149563.8X-3334,r=0.99997(见表1、图3);采用加样回收实验,结果表明:没食子酸的平均回收率为99.97%(n=6),RSD为0.51%(见表2);没食子酸的精密度(见表3);稳定性(见表4);重复性(见表5);专属性(见图4、5、6)与柱耐用性(见表6、图7、8、9);石榴皮止泻散中没食子酸含量结果(表7);提取溶剂对鞣花酸含量影响(表8);鞣花酸的进样量在0.01302~0.2083μg,与峰面积呈良好的线性关系,回归方程为:Y=13048189.2X-21214r=0.9998(见表9、图11);鞣花酸的平均回收率为99.86%(n=6),RSD为0.55%(见表10);鞣花酸的精密度(见表11);稳定性(见表12);重复性(见表13);专属性(见图12、13、14)与柱耐用性(见表14、图15、16、17);石榴皮止泻散中鞣花酸酸含量结果(表7);等实验数据均符合方法学要求。适用于石榴皮止泻散中没食子酸和鞣花酸的定量测定。
本发明解决其技术问题所采用的技术方案为:
(1)定性鉴别方法
取石榴皮止泻散0.5g,研细,加60%甲醇2ml,超声处理15分钟,上清液作为供试品溶液;再取石榴皮对照药材0.5g,同法制备对照药材溶液;吸取上述二种溶液各8~10μl,分别点于同一硅胶GF254薄层板上,以体积比为2.5∶5∶0.3∶0.2的环己烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,置紫外光灯254nm下检视,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色主斑点;
(2).总没食子酸含量测定方法
A.色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以体积比为3∶5.4∶91.6的乙腈-甲醇-0.1%磷酸为流动相;检测波长:215nm;柱温为30℃;理论板数按去没食子酸峰计算不低于2500;
B.对照品溶液制备 取没食子酸对照品适量,精密称定,加50%甲醇制成每1ml含2.5μg的溶液,即得;
C.供试品溶液制备 取通过3号筛的石榴皮止泻散粉末0.1g,精密称定,置具塞锥形瓶中,加水15ml和浓盐酸6ml,加热回流3小时,放冷,加20%氢氧化钠4ml,摇匀,用水定量转移至50ml量瓶中至刻度,摇匀,滤过,精密量取续滤液2ml,置10ml量瓶中,加50%甲醇至刻度,摇匀,滤过,取续滤液作为供试品溶液;
D.测定法 分别精密吸取对照品溶液和供试品溶液各20μl,注入液相色谱仪,测定,即得;
(3).鞣花酸含量测定方法
A.色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以体积比为18∶5∶77的乙腈-甲醇-0.1%磷酸为流动相;检测波长:254nm;柱温为30℃;理论板数按去鞣花酸峰计算不低于4000;
B.对照品溶液的制备 取鞣花酸对照品适量,加甲醇制成每1ml含2μg的溶液,即得;
C.供试品溶液的制备 取通过3号筛的石榴皮止泻散粉末0.1g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,以功率250w、频率33kHz,超声处理40分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液1ml,置10ml量瓶中,加80%甲醇至刻度,摇匀,用0.45μm的微孔滤膜滤过,取续滤液作为供试品溶液;
测定法 分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。
本发明的原理如下:
1.依据中药各有效成分的化学结构与性质,遵循相似相容的提取原则,采用适宜的提取溶剂,简便、快捷地制备供试品与对照药材溶液。再用不同极性的展开剂,进行展开,各种化学成分就会随着不同的展开剂,依据吸附、解吸附、再吸附、解吸附的能力不同,而在各自的薄层板上得以良好分离,供试品色谱图在与对照药材色谱相应的位置上呈现相同的薄层斑点,而得以鉴别。
2.依据没食子酸在215nm有最大吸收,而鞣花酸在254nm有最大吸收,通过流动相的组分与比例探讨,使没食子酸或鞣花酸波峰得以良好分离,且波峰对称、保留时间适宜,在3种不同的色谱柱上,没食子酸保留时间10分钟左右,鞣花酸保留时间13~15分钟。再依据该2种成分的进样量在一定范围内,与其峰面积呈现良好的线性关系,而用于定量测定。
本发明的创新点及有益效果如下:
(1)本发明薄层鉴别供试品与对照药材只一步2ml60%甲醇超声,上清液点样、展开,紫外光灯254nm下检视即可,无过滤、浓缩、蒸干、显色步骤,前处理时间15分钟,溶剂4ml;与药典方法相比,检测一批样品可节约样品前处理时间2.5小时,有机溶剂105ml,显色剂10ml。
(2)本发明的总没食子酸的含量测定方法,与已报道方法相比,在供试品的制备上,只需样品0.1g,酸性水为溶媒,水解3小时后,适量碱中和过高的酸性,用水直接定容于50ml、取2ml续滤液,用50%甲醇稀释至10ml,滤过,即可;而报道方法需样品 20g,酸水250ml、乙醚250ml,要重复水解2次,每次2小时,合并滤液,蒸干,甲醇定容于100ml,滤过,进样,即可。两法相比,本方法比报道法节约有机溶剂596ml、时间2.5小时;从检测灵敏度上,本发明供试品溶液的浓度是0.0004g/ml药材,而报道方法则是0.2g/ml药材,二者相差500倍,也就是报道方法进样的一次,等于本方法进样500次,本方法大幅度延长了仪器与色谱柱寿命。从方法准确性上,本方法测定的6批不同产地石榴皮生产的散剂,总没食子酸平均含量为9.08mg/g,而报道方法测定的13批石榴皮的平均含量为2.92mg/g,本方法数值是报道方法的3倍,理由是本法酸浓度高,水解在酸水一相溶剂中,均匀、完全。综上本发明简便、快捷、准确、成本低、灵敏度高、环保、无溶剂蒸发给环境带来的污染。
(3)与药典记载的石榴皮中鞣花酸的含量测定相比:
1)流动相不同,药典是乙腈-0.2%磷酸(21∶79),一则酸的浓度大,pH值在2.0以下,极易损坏仪器与色谱柱,二则鞣花酸的波峰拖尾,峰的对称性不好,修改为体积比为18∶5∶77的乙腈-甲醇-0.1%磷酸后,解决了鞣花酸的波峰拖尾,峰型对称,流动相的pH值也提高到2.0以上;
2)鞣花酸对照品进样量不同,药典进样量鞣花酸为0.1~0.2μg,本发明为0.04μg,峰面积仍在50万,完全可以满足准确测定含量的要求,药典进样量是本发明的2.5~5倍,波峰面积要达到百万到二百多万,试想高浓度进一针等于本研究进3针,对于色谱柱寿命讲,本研究可以得到延长;
3)提取溶剂不同,从鞣花酸流动相中有机相的比例在21%,说明其是一偏水溶性的化合物,在甲醇中的溶解度应比含水甲醇中低,所以经对比研究,采用80%甲醇提取,可使鞣花酸含量提高9.9%(见表8),使测定结果更接近样品的真实含量,进而提高了方法准确性。
附图说明
图1 石榴皮止泻散的薄层鉴别图
图2 没食子酸光谱扫描图
图3 没食子酸线性关系图
图4 没食子酸对照品HPLC色谱图
图5 石榴皮止泻散中没食子酸HPLC色谱图
图6 没食子酸空白样品HPLC色谱图
图7 没食子酸柱耐用性试验-岛津柱HPLC色谱图
图8 没食子酸柱耐用性试验-luna柱HPLC色谱图
图9 没食子酸柱耐用性试验-安捷伦柱HPLC色谱图
图10 鞣花酸光谱扫描图
图11 鞣花酸线性关系图
图12 鞣花酸对照品HPLC色谱图
图13 石榴皮止泻散中鞣花酸HPLC色谱图
图14 鞣花酸空白样品HPLC色谱图
图15 鞣花酸柱耐用性试验-岛津-VP-ODS柱HPLC色谱图
图16 鞣花酸柱耐用性试验-luna柱HPLC色谱图
图17 鞣花酸柱耐用性试验-月旭柱HPLC色谱图
图1中,1.2.3为样品,4.为石榴皮对照药材。
图2、图10中,横坐标为波长(nm)、纵坐标为吸收度(mAU)。
图3、图11中,横坐标为进样量(μg)、纵坐标为峰面积。
图4、图5、图6、图7、图8、图9中,横坐标为保留时间(分钟),纵坐标为响应值(uV),1为没食子酸峰。
图12、图13、图14、图15、图16、图17中,横坐标为保留时间(分钟),纵坐标为响应值(uV),1为鞣花酸峰。
本发明具体实施方式
实施例
(1)定性鉴别方法
取石榴皮止泻散0.5g,研细,加60%甲醇2ml,超声处理20分钟,上清液作为供试品溶液。再取石榴皮对照药材0.5g,同法制备对照药材溶液。吸取上述二种溶液各8~10μl,分别点于同一硅胶GF254薄层板上,以体积比为2.5∶5∶0.3∶0.2的环己烷-乙酸乙酯-甲醇-甲酸为展开剂,展开,取出,晾干,置紫外光灯254nm下检视;供试品色谱中,在与对照品色谱相应的位置上,显相同颜色主斑点;
(2).总没食子酸含量测定方法
A.色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以体积比为3∶5.4∶91.6的乙腈-甲醇-0.1%磷酸为流动相;检测波长:215nm;柱温为30℃;理论板数按去没食子酸峰计算不低于2500;
B.对照品溶液制备 取没食子酸对照品适量,精密称定,加50%甲醇制成每1ml含2.5μg的溶液,即得;
C.供试品溶液制备取通过3号筛的石榴皮止泻散粉末0.1g,精密称定,置具塞锥形瓶中,加水15ml和浓盐酸6ml,加热回流3小时,放冷,加20%氢氧化钠4ml,摇匀,用水定量转移至50ml量瓶中至刻度,摇匀,滤过,精密量取续滤液2ml,置10ml量瓶中,加50%甲醇至刻度,摇匀,滤过,取续滤液作为供试品溶液;
D.测定法 分别精密吸取对照品溶液和供试品溶液各20μl,注入液相色谱仪,测定,即得,测定了6批样品含量(见表7);
(3).鞣花酸含量测定方法
A.色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以体积比为18∶5∶77的乙腈-甲醇-0.1%磷酸为流动相;检测波长:254nm;柱温为30℃;理论板数按去鞣花酸峰计算不低于4000;
B.对照品溶液的制备 取鞣花酸对照品适量,加甲醇制成每1ml含2μg的溶液,即得;
C.供试品溶液的制备 取通过3号筛的石榴皮止泻散粉末0.1g,精密称定,置具塞锥形瓶中,精密加入80%甲醇25ml,密塞,称定重量,以功率250w、频率33kHz,超声处理40分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液1ml,置10ml量瓶中,加80%甲醇至刻度,摇匀,用0.45μm的微孔滤膜滤过,取续滤液作为供试品溶液;
测定法 分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得,测定了6批样品含量(见表7);
表1 没食子酸进样量与峰面积
表2 样品中没食子酸的回收率试验结果
表3 没食子酸精密度实验结果(峰面积)
表4 没食子酸稳定性实验结果
表5 没食子酸重复性试验结果
表6 没食子酸柱耐用性试验结果(mg/g)
表7 石榴皮止泻散中没食子酸含量测定结果
表8 不同浓度甲醇提取对鞣花酸含量影响(mg/g)
表9 鞣花酸进样量与峰面积
表10 样品中鞣花酸的回收率试验结果
表11 鞣花酸精密度实验结果(峰面积)
表12 鞣花酸稳定性实验结果
表13 鞣花酸重复性试验结果
表14 鞣花酸柱耐用性试验结果(mg/g)
参考文献
[1]张朔生,HPLC法测定炮制前后石榴皮中没食子酸含量,药物分析杂志,[J].2010.30(6)1104~1106
[2]周本宏,刘春,吴振华等,高效液相色谱法测定石榴皮中没食子酸的含量,[J].中国医院药学杂志,2005.25(12)1148~1150
[3]常占瑛,马桂芝,高晓黎,HPLC法测定新疆不同产地石榴皮中没食子酸的含量,[J].新疆医科大学学报.2009.32(11)1552~1553。
- 还没有人留言评论。精彩留言会获得点赞!