一种平贝母药材指纹图谱的建立方法与流程
本发明涉及一种中药药材的质量控制方法,特别涉及平贝母药材高效液相色谱法指纹图谱的建立方法。
背景技术:
平贝母为百合科贝母属多年生草本植物平贝母(Fritillaria ussurienis Maxim)的干燥鳞茎,又名平贝,是药用贝母的一种,收载于2010版《中国药典》及香港中药材标准第三期(2010年)。
平贝母主产东北三省,性苦、甘,微寒;归肺、心经。早在秦汉时期《神农本草经》将贝母列为中品,能清热润肺,化痰止咳,用于肺热燥咳、干咳少痰、阴虚劳咳、咳痰带血。
甾体生物碱成分是现有文献报道的公认的平贝母药材的主要活性成分,也是作为平贝母清热润肺、化痰止咳的物质基础。目前中国药典(2010年)控制平贝母药材质量的方法主要包括性状鉴定、薄层鉴别及含量测定等方法。含量测定主要通过利用分光光度法测定平贝母药材中总生物碱含量来达到控制平贝母药材质量的目的。而香港中药材标准第三期(2010年)还收载了平贝母的指纹图谱,但其方法中供试品制备取样量大,因使用三氯甲烷为提取溶剂且量多,毒性大,指纹图谱特征峰较少。闵会等研究了不同贝母的指纹图谱鉴别研究,指纹图谱只分离出了7个特征峰,对于药材中可能存在的其他共同特征峰或许没有完全被检测。
贝母辛、贝母素甲、贝母素乙、西贝母碱为平贝母中主要的生物碱,由于它们缺少发色团,无紫外吸收,因此不能用紫外吸收检测器检测。研究者用薄层扫描法、柱前衍生化法、质谱液相联用法、气相色谱法测定平贝母中这类生物碱含量,但多因检测的局限性难以得到理想的结果。近年,使用高效液相色谱法检测平贝母中生物碱成分的研究较多,李慧婷等人用ELSD-HPLC测定平贝母中贝母素甲、贝母素乙含量,吴晓民等人用HPLC测定平贝母中贝母甲素的含量,洪梅等人用ELSD-HPLC等度洗脱测定平贝母中贝母素甲的含量,这些方法只能测定一种或两种生物碱成分,并不能完全反映药材的整体特征。王世曼等人曾用HPLC研究吉林产贝母生物碱不同提取方法的指纹图谱,以贝母素乙为参照,但经查阅文献,贝母素乙在平贝母中含量极低,以贝母素乙为参照会对准确性造成一定的影响。
技术实现要素:
本发明涉及平贝母药材高效液相色谱法指纹图谱的建立方法,以及由此方法得到的平贝母药材的标准指纹图谱。通过测定平贝母药材的指纹图谱有效地控制平贝母药材的质 量,弥补了现有质量控制技术的不足,从而确保平贝母药材的有效性、稳定性和均一性。
本发明通过以下技术方案实现上述目的:一种平贝母药材的指纹图谱,该方法包括采用高效液相色谱法对平贝母进行检测,其中色谱条件包括;
色谱柱:十八烷基硅烷键合硅胶为填充剂;
流动相:流动相A为含1mmol/L乙酸铵的0.1%三乙胺水溶液,流动相B为乙腈溶液;
采用梯度洗脱,洗脱步骤如下,其中流动相比例均为体积百分比:
0~8min,流动相A为70%~65%,流动相B为30%~35%;
8~25min,流动相A为65%~55%,流动相B为35%~45%;
25~40min,流动相A为55%~5%,流动相B为45%~95%;
40~43min,流动相A为5%,流动相B为95%;
43~48min,流动相A为5%~70%,流动相B为95%~30%;
48~58min,流动相A为70%,流动相B为30%。所述平贝母指纹图谱的建立方法,
还包括通过以下步骤制备对照品溶液:
取贝母辛、贝母素甲、贝母素乙、西贝母碱四种生物碱对照品适量,精密称定,加甲醇制成每ml含贝母辛0.140mg、贝母素甲0.133mg、贝母素乙0.100mg、西贝母碱0.372mg的混合对照品溶液,即得。
所述平贝母指纹图谱的建立方法,还包括通过以下步骤制备供试品溶液:把平贝母打粉,过四号筛,取2g平贝母药材粉末,加氨水4ml浸润2h,用比例为4:1的三氯甲烷-甲醇混合溶液40ml回流提取,过滤。取滤液,蒸干,残渣用甲醇溶解定容至2ml,12000r/min离心10min沉淀不溶性物质,取上清液,将上清液与1g硅胶混合均匀,通过内径为1.5cm、填料重量为5g的薄层层析硅胶柱,以水30ml洗脱,弃去水洗液,继用比例为9:1的乙酸乙酯-甲醇40ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解定容至2ml,用0.22μm微孔滤膜滤过,即得。
优选地,所述平贝母指纹图谱的建立方法,包括以下步骤:
(a)制备供试品溶液:把平贝母打粉,过四号筛,取2g平贝母药材粉末,加氨水4ml浸润2h,用比例为4:1的三氯甲烷-甲醇混合溶液40ml回流提取,过滤。取滤液,蒸干,残渣用甲醇溶解定容至2ml,12000r/min离心10min沉淀不溶性物质,取上清液,将上清液与1g硅胶混合均匀,通过内径为1.5cm、填料重量为5g的薄层层析硅胶柱,以水30ml洗脱,弃去水洗液,继用比例为9:1的乙酸乙酯-甲醇40ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解定容至2ml,用0.22μm微孔滤膜滤过,即得;
(b)制备对照品溶液:取贝母辛、贝母素甲、贝母素乙、西贝母碱四种生物碱对照品适量,精密称定,加甲醇制成每ml含贝母辛0.140mg、贝母素甲0.133mg、贝母素乙 0.100mg、西贝母碱0.372mg的混合对照品溶液,即得;
(c)测定:精密吸取对照品溶液与供试品溶液各10μl注入高效液相色谱仪,根据以下条件进行测定,得到平贝母药材指纹图谱;
其中,色谱条件包括:
色谱柱:十八烷基硅烷键合硅胶为填充剂;
流动相:流动相A为含1mmol/L乙酸铵的0.1%三乙胺水溶液,流动相B为乙腈溶液;
采用梯度洗脱,洗脱步骤如下,其中流动相比例均为体积百分比:
0~8min,流动相A为70%~65%,流动相B为30%~35%;
8~25min,流动相A为65%~55%,流动相B为35%~45%;
25~40min,流动相A为55%~5%,流动相B为45%~95%;
40~43min,流动相A为5%,流动相B为95%;
43~48min,流动相A为5%~70%,流动相B为95%~30%;
48~58min,流动相A为70%,流动相B为30%;流速为1ml/min;
柱温为25℃;
进样量为10μl;蒸发光散射器的条件包括:
漂移管温度:85℃;雾化器流速:2.2L/min;
Grain:1。所述平贝母指纹图谱的建立方法,还包括:对多个平贝母指纹图谱进行比较,选出共有特征峰,得到平贝母特征指纹图谱。所述平贝母指纹图谱的建立方法,在于平贝母指纹图谱或平贝母特征指纹图谱中,以贝母辛对照品为参照峰,各特征峰的相对保留时间如下:峰1:0.79、峰2:0.97、峰3:1.00、峰4:1.32、峰5:1.51、峰6:1.83、峰7:2.10、峰8:2.21、峰9:2.28、峰10:2.32、峰11:2.43、峰12:2.59、峰13:2.62、峰14:2.74,其相对保留时间在上述值的±10%之内。所述平贝母指纹图谱或平贝母特征指纹图谱中,3号峰为贝母辛对照峰。本发明还提供一种平贝母药材的鉴定方法,该方法包括将根据上述方法建立待测样品指纹图谱或特征指纹图谱与根据上述方法建立的标准指纹图谱或特征指纹图进行比较。
所述平贝母药材的鉴定方法,包括以下步骤:
(a)测定待测平贝母药材生物碱类指纹图谱;
(b)将待测平贝母药材指纹图谱与标准指纹图谱对比,以特征峰计算,相似度不低于0.85的药材质量合格,其中所述标准指纹图谱包括14个共有峰,其中3号峰为贝母辛 对照峰,各对照峰的相对保留时间为:峰1:0.79、峰2:0.97、峰3:1.00、峰4:1.32、峰5:1.51、峰6:1.83、峰7:2.10、峰8:2.21、峰9:2.28、峰10:2.32、峰11:2.43、峰12:2.59、峰13:2.62、峰14:2.74,其相对保留时间在上述值的±10%之内。
不同色谱条件的对比
1、色谱柱:比较C18柱、C8柱、苯基柱、氨基柱等常用色谱柱,发现C8柱与苯基柱对平贝母有药材中成分保留极差,不能全面反映平贝母药材成分。氨基柱为正向分配体系,不适合本次实验。本方法选择C18柱进行分离,得到了较好的效果。
2、流动相:单纯使用乙腈-三乙胺水体系或者甲醇-三乙胺水体系,无法将药品中的成分尽可能的分离,且峰形难看,保留时间波动较大,从而很难依据其图谱进行鉴别,根本不能满足特征图谱的建立要求。本方法结合现有技术的流动相及其分离的平贝母中生物碱的峰的特点,最终确定使用乙腈-三乙胺-乙酸铵的三元体系,此条件下,各色谱峰峰形尖锐,分离度较好,保留时间适中。
3、检测器:平贝母中生物碱成分属于甾体类生物碱,无紫外吸收,且单一的有效成分含量极低,不能适宜选择常用的紫外检测器,本方法选用灵敏度高的蒸发光散色检测器,检测条件为,漂移管温度:85℃;雾化器流速:2.2L/min;Grain:1。
本发明将平贝母药材作为一个整体对待,通过比较其特征峰可以找出不同药材之间的细微差异,适用于平贝母药材真伪、产地、品质的鉴别和控制。
与现有技术相比,本发明的优点是:
1、现行《中国药典》(2010年)标准中,对平贝母的鉴别方法只有薄层鉴定和总生物碱含量鉴定,本发明所述平贝母药材的鉴定方法简便、重现性好,准确率高,只要供选择的药材中出现本申请所指定的指纹图谱,即可确认药材为平贝母药材,确保了药材选择的准确性,确保了成药的质量。
2、本发明采用乙腈-三乙胺-乙酸铵系统,利用梯度洗脱的方法建立了平贝母药材生物碱类指纹图谱,利用乙酸铵屏蔽硅醇基改善峰形,三乙胺调节峰拖尾。该方法操作简便,稳定可靠,精密度高,分离度好。
3、平贝母药材多以总生物碱为质量控制指标,但并不能完成反映中药的质量。近年来,中药指纹图谱和特征指纹图谱已广泛应用于质量控制,我们运用指纹图谱技术有效地控制平贝母药材质量,弥补了现有质量控制技术的不足,从而确保平贝母药材的有效性、稳定性和均一性。
4、生物碱类成分试现有文献报道的公认的平贝母药材的主要活性成分,文献报道测定平贝母中生物碱类成分只能测定一种或两种成分,平贝母共有峰一般被检测出6-7个共有峰,本发明在测定平贝母生物碱类成分中,同时测定平贝母中贝母辛、贝母素甲、贝母 素乙、西贝母碱四种有效生物碱成分。
5、本发明所述平贝母药材的鉴定方法中,还包括对该药材的供试品制备方法的步骤,首先利用传统的方法氨水浸润、氯仿-甲醇提取后,继用薄层层析硅胶吸附,水溶液洗脱除去水溶性物质,再用乙酸乙酯-甲醇液将生物碱成分洗脱,减少杂质对液相测定的影响。
6、本发明具有方法简便,易于掌握,稳定,精密度高,重现性好的特点。
附图说明
图1为实施例1平贝母药材生物碱类成分标准指纹图谱
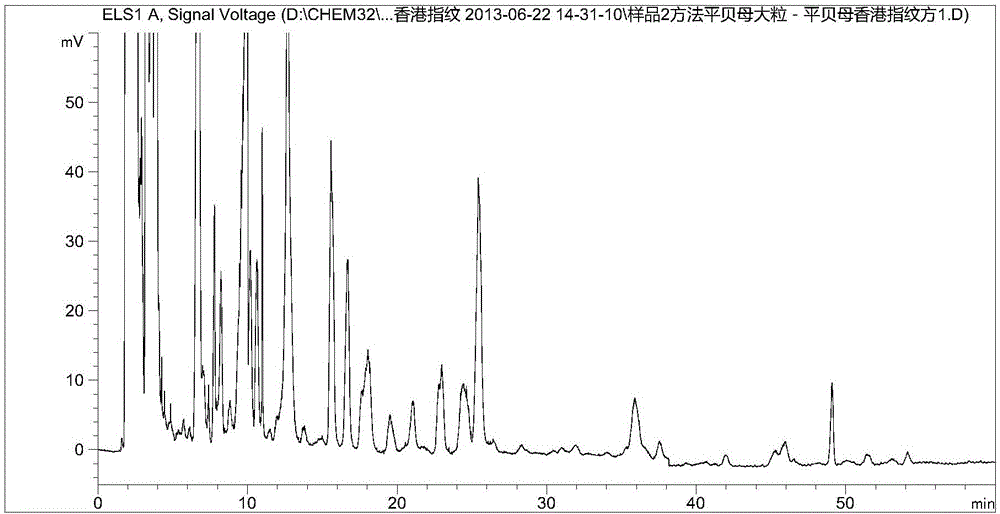
图2为对比例1中所述平贝母药材特征峰的指纹图谱—香港指纹图谱方法
图3为对比例2中所述平贝母药材特征峰的指纹图谱—参考文献方法
图4为实施例2十批平贝母药材样品生物碱类成分指纹图谱
具体实施方式
下面通过实施例进一步说明本发明。应该理解的是,本发明的实施例是用于说明本发明而不是对本发明的限制。根据本发明的实质对本发明进行的简单改进都属于本发明要求保护的范围。
实施例1:平贝母药材生物碱类成分指纹图谱建立实验
1、仪器、试药和供试样品
仪器:高效液相色谱仪:Aglient1200,蒸发光散色器:Alltech 3300;
电子天平:型号:CP214,奥豪斯仪器上海有限公司制造;
对照品:贝母辛对照品(中国食品药品检定研究,批号:11892-201201);贝母素甲对照品(中国食品药品检定研究,含量为98.3%,批号:110750-201110);贝母素乙对照品(中国食品药品检定研究,含量为99.9%,批号:110751-201110);西贝母碱对照品(中国食品药品检定研究,含量为99.1%,批号:110767-201107)。
样品:平贝母药材(梧州制药有限公司采购部供应,批号:13082101、13082102、13082103、13082104、13082105、13082106、13082107、13082108、13082109、13082110)。
2、液相色谱条件
以十八烷基硅烷键合硅胶为填充剂,流动相A为0.1%三乙胺(含1mmol/L的乙酸铵)的水溶液,流动相B为乙腈溶液;流速为1.0ml/min,采用梯度洗脱,洗脱程序如下,
0~8min,流动相A为70%~65%,流动相B为30%~35%;
8~25min,流动相A为65%~55%,流动相B为35%~45%;
25~40min,流动相A为55%~5%,流动相B为45%~95%;
40~43min,流动相A为5%,流动相B为95%;
43~48min,流动相A为5%~70%,流动相B为95%~30%;
48~58min,流动相A为70%,流动相B为30%;
流速为1ml/min;
柱温为25℃;
进样量为10μl;
3、蒸发光散射器的条件:
漂移管温度:85℃;
雾化器流速:2.2L/min;
Grain:1。
4、对照品溶液的制备
取贝母辛、贝母素甲、贝母素乙、西贝母碱四种生物碱对照品适量,精密称定,加甲醇制成每ml含贝母辛0.140mg、贝母素甲0.133mg、贝母素乙0.100mg、西贝母碱0.372mg的混合对照品溶液,即得。
5、供试品溶液的制备
把平贝母打粉,过四号筛,取2g平贝母药材粉末,加氨水4ml浸润2h,用三氯甲烷-甲醇(4:1)混合溶液40ml回流提取,过滤。取滤液,蒸干,残渣用甲醇溶解定容至2ml,12000r/min离心10min沉淀不溶性物质,取上清液,将上清液与1g硅胶混合均匀,通过薄层层析硅胶柱(内径为1.5cm,5g),以水30ml洗脱,弃去水洗液,继用乙酸乙酯-甲醇(9:1)40ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解定容至2ml,用0.45μm微孔滤膜滤过,即得。
6、平贝母药材标准指纹图谱的制定
取平贝母药材10批,按照供试品溶液的制备方法制备供试品,在上述液相色谱条件下,分别精密吸取参照物溶液与供试品溶液各10μl,注入液相色谱仪,记录色谱图。依据10批样品的指纹图谱,制定特征图谱。供试品特征图中应呈现14个特征峰,其中4个峰应与相应的对照品峰保留时间相一致;以贝母辛参照峰为S峰,计算各特征峰与S峰的相对保留时间,结果表明,各共有特征峰的相对保留时间的RSD均小于2%,符合指纹图谱分析要求。根据标准指纹图谱,相对保留时间规定值为峰1:0.79、峰2:0.97、峰3:1.00、峰4:1.32、峰5:1.51、峰6:1.83、峰7:2.10、峰8:2.21、峰9:2.28、峰10:2.32、峰11:2.43、峰12:2.59、峰13:2.62、峰14:2.74。在14个特征峰中,3号峰与贝母辛对应的峰,14号峰与西贝母碱,其保留时间一致。
表1共有特征峰相对保留时间表
对比例1:平贝母药材生物碱类成分指纹图谱建立实验
1、仪器、试药和供试样品
仪器:高效液相色谱仪:Aglient1200,蒸发光散色器:Alltech 3300;
电子天平:型号:CP214,奥豪斯仪器上海有限公司制造;
对照品:贝母辛对照品(中国食品药品检定研究,批号:11892-201201);贝母素甲对照品(中国食品药品检定研究,含量为98.3%,批号:110750-201110);贝母素乙对照品(中国食品药品检定研究,含量为99.9%,批号:110751-201110);西贝母碱对照品(中国食品药品检定研究,含量为99.1%,批号:110767-201107)。
样品:平贝母药材(梧州制药有限公司采购部供应,批号:13082101、13082102、13082103)。
2、液相色谱条件
以十八烷基硅烷键合硅胶为填充剂,流动相A为0.1%三乙胺溶液,流动相B为乙腈溶液;流速为1.0ml/min,采用梯度洗脱,洗脱程序如下,
0~10min,流动相A为70%~65%,流动相B为30%~35%;
10~32min,流动相A为65%~56%,流动相B为35%~44%;
32~60min,流动相A为56%~45%,流动相B为44%~55%;
蒸发光散色检测器,检测条件为漂移管温度:105℃;雾化器流速(N2):2.8L/min;Grain:1。
3、对照品溶液的制备取贝母辛、贝母素甲、贝母素乙、西贝母碱四种生物碱对照品适量,精密称定,加甲醇制成每ml含贝母辛0.140mg、贝母素甲0.133mg、贝母素乙0.100mg、西贝母碱0.372mg的混合对照品溶液,即得。
4、供试品溶液的制备
把平贝母打粉,过四号筛,取5g平贝母药材粉末,加氨水10ml浸润1h,加二氯甲烷100ml,加热回流2小时,放冷至室温,过滤。取滤液,滤液转移与200ml圆底烧瓶中,用旋转蒸发器减压蒸干。残渣溶于甲醇,转移与2ml量瓶中,加甲醇至刻度,用0.45μm微孔滤膜滤过,即得。
5、平贝母药材标准指纹图谱的制定
在上述液相色谱条件下,分别精密吸取参照物溶液与供试品溶液各10μl,注入液相色谱仪,记录色谱图。供试品特征图中特征峰少,多数集中在30分钟前,30分钟后无显著的特征峰,主要检测出贝母辛成分。
图2为对比例1中所述平贝母药材特征峰的指纹图谱—香港指纹图谱方法。
对比例2:平贝母药材生物碱类成分指纹图谱建立实验
1、仪器、试药和供试样品
仪器:高效液相色谱仪:Aglient1200,蒸发光散色器:Alltech 3300;
电子天平:型号:CP214,奥豪斯仪器上海有限公司制造;
对照品:贝母辛对照品(中国食品药品检定研究,批号:11892-201201);贝母素甲对照品(中国食品药品检定研究,含量为98.3%,批号:110750-201110);贝母素乙对照品(中国食品药品检定研究,含量为99.9%,批号:110751-201110);西贝母碱对照品(中国食品药品检定研究,含量为99.1%,批号:110767-201107)。
样品:平贝母药材(梧州制药有限公司采购部供应,批号:13082101、13082102、13082103)。
2、液相色谱条件
以十八烷基硅烷键合硅胶为填充剂,流动相A为水溶液,流动相B为甲醇溶液(含0.05%三乙胺);流速为1.0ml/min,采用梯度洗脱,洗脱程序如下,
0~10min,流动相A为40%~30%,流动相B为60%~70%;
10~35min,流动相A为30%~10%,流动相B为70%~90%;
35~50min,流动相A为10%~0%,流动相B为90%~100%;
50~60min,流动相A为0%,流动相B为100%;
蒸发光散色检测器,检测条件为漂移管温度:70℃;雾化器流速(N2):1.6L/min。
3、对照品溶液的制备
取贝母辛、贝母素甲、贝母素乙、西贝母碱四种生物碱对照品适量,精密称定,加甲醇制成每ml含贝母辛0.140mg、贝母素甲0.133mg、贝母素乙0.100mg、西贝母碱0.372mg的混合对照品溶液,即得。
4、供试品溶液的制备
取2g平贝母药材粉末,加氨水4ml浸润1h,用三氯甲烷-甲醇(4:1)混合溶液40ml回流提取2h,放冷,精密加入6ml无水乙醇,用力摇匀,精密量取续滤液20ml,水浴蒸干,残渣加甲醇5ml溶解,滤过,即得。
5、平贝母药材标准指纹图谱的制定
在上述液相色谱条件下,分别精密吸取参照物溶液与供试品溶液各10μl,注入液相色谱仪,记录色谱图。供试品特征图中特征峰保留时间长,在29.78min、40.75min出现,特征峰只检测出贝母辛和西贝母碱两种成分,41分钟后无显著的特征峰。
图3为对比例2中所述平贝母药材特征峰的指纹图谱—参考文献方法。
实施例2:平贝母药材HPLC特征图谱建立:
平贝母药材由广西梧州制药(集团)股份有限公司采购部提供。
照高效液相色谱法(《中国药典》2010年版一部附录VID)测定:
以十八烷基硅烷键合硅胶为填充剂,流动相A为0.1%三乙胺(含1mmol/L的乙酸铵)的水溶液,流动相B为乙腈溶液;流速为1.0ml/min,采用梯度洗脱,洗脱程序如下,
0~8min,流动相A为70%~65%,流动相B为30%~35%;
8~25min,流动相A为65%~55%,流动相B为35%~45%;
25~40min,流动相A为55%~5%,流动相B为45%~95%;
40~43min,流动相A为5%,流动相B为95%;
43~48min,流动相A为5%~70%,流动相B为95%~30%
48~58min,流动相A为70%,流动相B为30%
对照品溶液的制备:取贝母辛、贝母素甲、贝母素乙、西贝母碱四种生物碱对照品适 量,精密称定,加甲醇制成每ml含贝母辛0.140mg、贝母素甲0.133mg、贝母素乙0.100每个、西贝母碱0.372每个的混合对照品溶液,即得。
供试品溶液的制备:把平贝母打粉,过四号筛,取2g平贝母药材粉末,加氨水4ml浸润2h,用三氯甲烷-甲醇(4:1)混合溶液40ml回流提取,过滤。取滤液,蒸干,残渣用甲醇溶解定容至2ml,12000r/min离心10min沉淀不溶性物质,取上清液,将上清液与1g硅胶混合均匀,通过薄层层析硅胶柱(内径为1.5cm,5g),以水30ml洗脱,弃去水洗液,继用乙酸乙酯-甲醇(9:1)40ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解定容至2ml,用0.22μm微孔滤膜滤过,即得。
测定法:分精密吸取对照品溶液与供试品溶液各10μl注入高效液相色谱仪,测定,记录色谱图,即得:
供试品特征图谱中应呈现14个特征峰,其中峰3和峰14分别与相应的对照品峰保留时间一致,以贝母辛对照品参照峰为S峰,计算各特征峰与S峰的相对保留时间,结果表明,各共有特征峰的相对保留时间的RSD均小于2%,符合指纹图谱分析要求。根据标准指纹图谱,相对保留时间规定值为峰1:0.79、峰2:0.97、峰3:1.00、峰4:1.32、峰5:1.51、峰6:1.83、峰7:2.10、峰8:2.21、峰9:2.28、峰10:2.32、峰11:2.43、峰12:2.59、峰13:2.62、峰14:2.74,其相对保留时间在上述值的±10%之内。
- 还没有人留言评论。精彩留言会获得点赞!