一种阿魏酸钠、阿司匹林、桂利嗪和维生素B1的含量测定方法与流程
本发明涉及一种阿魏酸钠、阿司匹林、桂利嗪和维生素B1的含量测定方法,具体涉及复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、阿司匹林、桂利嗪和维生素B1的含量测定方法。
背景技术:
复方阿魏酸钠阿司匹林胶囊是丽珠集团利民制药厂研制的西药第3类新药(每粒含50mg阿魏酸钠,20mg阿司匹林,桂利嗪25mg和10mg维生素B1),本品的4种成分阿魏酸钠、桂利嗪、阿司匹林和维生素B1在抗血小板聚集、抗凝、降低血粘度、溶栓、扩血管、维护心脑血管正常功能方面具有不同的作用机制,经组方有协同作用,使本品具有较强的抗血小板聚集,有明显抑制实验性血栓形成和较强的抗实验性血栓的作用,适用于预防和治疗缺血性脑血管疾病,如短暂脑缺血性发作、脑缺血性中风和脑动脉硬化,其组方科学生物利用度高疗效确切用药安全,有很好的市场效应。但是由于其组方较为复杂,且各成分均为微量用药,现行质量标准——国家药品标准(新药转正标准)第45册各组分采用单一方法独立检验,分别采用UV法、UV法、重量分析法测定,各组分检验前处理复杂,可操作的重复差、难度高、准确度低、检验周期长,不利于产品的质量控制及延误生产进度,检验成本高等的缺陷。
现有文献公开了用HPLC法单独检测固体制剂中阿魏酸钠、阿司匹林、桂利嗪和维生素B1,如顾玉英等公开的《HPLC法测定定痛宁冲剂中阿魏酸的含量》采用Diamonsil C18色谱柱,甲醇∶1%醋酸溶液(30∶70)作为流动相,供试品溶液的制备方法:用水溶解后超声提取,然后三氯甲烷萃取,萃取液蒸干后残渣用甲醇溶解。郑维兵等《HPLC法测定归芪生血颗粒中阿魏酸的含量》采用Dimasoil C18色谱柱,乙腈-0.085%磷酸溶液(13∶87)作为流动相,供试品溶液的制备方法:用甲醇溶解超声,水浴,用醋酸乙酯萃取后蒸干,残渣用甲醇溶解。俞永梅等公开的《HPLC法测定参芪生肌颗粒中阿魏酸的含量》采用KromasilC18色谱柱,甲醇-0.1%磷酸溶液(30∶70)为流动相,供试品溶液的制备方法:用甲醇溶解过滤后即得。杨春艳等公开的《高效液相色谱法测定阿咖片中阿司匹林含量》采用Symmetry C18柱,磷酸盐缓冲液-甲醇(7∶3)为流动相,供试品溶液的制备方法:供试品磨碎,用流动相溶解过滤后即得。从以上公开文献可以看出,阿魏酸钠、阿司匹林、桂利嗪和维生素B1每种物质使用的色谱柱种类、流动相组成和供试品制备方法都不相同,阿魏酸钠和维生素B1水溶性极强,而阿司匹林和桂利嗪是水不溶性,若四种物质用同一HPLC条件测定含量,必将会遇到诸多问题。
蓝献泉等公开的《HPLC法同时测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、桂利嗪、阿司匹林、维生素B1》中,公开了HPLC一次性检测四种成分的方法,节约了时间,但是文中所提到的用初始比例流动相甲醇-乙腈-醋酸水(25∶20∶55)溶解样品超声过程中会使样品的包衣聚团导致样品中的桂利嗪难以溶解,含量检测结果远远低于生产理论值和重量法测定结果;同时该文献中供试品溶液制备方法:供试品研细,混合均匀,精密称取粉末适量,加初始比例流动相适量,超声过滤,因流动相中含有55%水相且超声长达15分钟,会导致样品中的阿司匹林快速水解产生大量的水杨酸,会导致样品中的阿司匹林低于标准要求,综上所述蓝献泉等公开方法并不适用于复方阿魏酸钠阿司匹林胶囊中的桂利嗪、阿司匹林的含量检测。
因此需要一种更加稳定的方法同时测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、桂利嗪、阿司匹林。
技术实现要素:
本方法涉及同一色谱条件测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、阿司匹林、桂利嗪组分含量的方法。
本发明所述方法还可以同时测定复方阿魏酸钠阿司匹林胶囊中维生素B1含量。
一种复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、阿司匹林、桂利嗪的含量测定方法,所述方法包括:
(1)色谱条件:以十八烷基硅烷键合硅胶为填充剂.流动相有A相和B相;其中A相为乙腈、四氢呋喃、冰醋酸和水的混合溶剂,B相为甲醇、三乙醇胺和三乙胺的混合溶剂,B相pH值为6.0-7.0;
(2)供试品溶液制备:取复方阿魏酸钠阿司匹林胶囊内容物置研砵,研细,称取约相当于阿魏酸钠50mg、阿司匹林20mg、桂利嗪25mg的细粉置量瓶中,加冰醋酸甲醇溶液适量,超声振荡,稀释至刻度,摇匀,滤过,作为供试品溶液;
(3)混合对照品溶液制备:精密称取阿魏酸钠对照品约50mg,阿司匹林对照品约20mg,桂利嗪对照品约25mg置量瓶中,加冰醋酸甲醇溶液超声溶解,并稀释至刻度,摇匀,作为对照品溶液;
(4)阴性对照品溶液制备:取混合辅料空白,精密称加,加冰醋酸甲醇溶液超声溶解,并稀释至刻度,摇匀,作为阴性对照品溶液;
(5)测定方法:分别吸取供试品溶液、混合对照品溶液、阴性对照品溶液各10ul,注入液相色谱仪,测定,记录色谱图,以峰面积按外标法计算各组分标示百分含量,即得。
优选的,所述步骤(1)中A相乙腈、四氢呋喃、冰醋酸和水的比例为20∶5∶5∶70。
优选的,所述步骤(1)中B相为甲醇、0.2%三乙醇胺和三乙胺混合溶剂,甲醇、0.2%三乙醇胺和三乙胺的比例为80∶20∶0.4。
优选的,所述步骤(1)中A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%。
优选的,所述步骤(1)中还包括:流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
优选的,所述步骤(2)-(4)中使用的冰醋酸甲醇溶液中冰醋酸的浓度为0.5%-5.0%。
优选的,所述步骤(2)-(4)中使用的冰醋酸甲醇溶液中冰醋酸的浓度为1.0%。
现行方法如国家药品标准(新药转正标准)中,使用组分分别测定的方法,测定其阿魏酸钠、阿司匹林、桂利嗪的含量需要三个工作天的时间,本发明涉及的方法只需一个工作天即可完成三个组分的含量测定,本发明涉及的方法样品处理一步到位,比现行方法减少了繁杂的样品处理过程,能较大的降低因现行方法繁杂的样品处理引起的系统误差及偶然误差,执行本发明涉及的方法后能更好控制复方阿魏酸钠阿司匹林胶囊的质量。
本发明涉及的方法能减少两个工作天的检验人力成本,同时缩短了中间产品检验周期,减少了车间中间产品到成品的等待时间,节约了时间成本及因等待而引起的能耗成本,获得一定的经济效益。
本发明涉及的方法含量测定过程中专属性高、重复性好、准确度高、精密度良好等的优异处。
本方法涉及同一色谱条件测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、阿司匹林、桂利嗪和维生素B1组分含量的方法。
一种复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、阿司匹林、桂利嗪和维生素B1的含量测定方法,所述方法包括:
(1)色谱条件:以十八烷基硅烷键合硅胶为填充剂;流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul;
(2)供试品溶液制备:临用新配,取复方阿魏酸钠阿司匹林胶囊20粒内容物置研砵,研细,精密称取约相当于阿魏酸钠50mg、阿司匹林20mg、桂利嗪25mg及10mg维生素B1的细粉(约相当于一粒的量)置100ml量瓶中,加1%冰醋酸甲醇溶液适量,超声2分钟使溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
(3)混合对照品溶液制备:临用新配,精密称取阿魏酸钠对照品约50mg,阿司匹林对照品约20mg,桂利嗪对照品约25mg及10mg维生素B1同置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为对照品溶液。
(4)阴性对照品溶液制备:取混合辅料空白,精密称取约1粒的量,同法制备,作为阴性对照品溶液。
(5)测定方法:分别吸取供试品溶液、混合对照品溶液、阴性对照品溶液各10ul,注入液相色谱仪,测定,记录色谱图,以峰面积按外标法计算各组分标示百分含量,即得。
附图说明
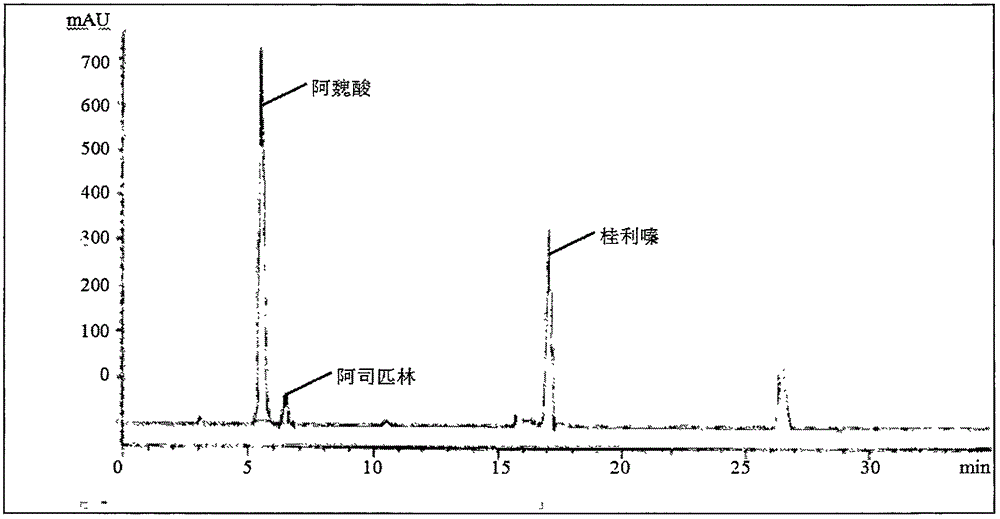
附图1:实施例2实验组D复方阿魏酸钠阿司匹林胶囊中维生素B1、阿魏酸钠、阿司匹林、桂利嗪测定色谱图;
附图2:实施例2实验组E复方阿魏酸钠阿司匹林胶囊中维生素B1、阿魏酸钠、阿司匹林、桂利嗪测定色谱图;
附图3:实施例5方法学验证专属性试验测定色谱图。
附图4:实施例5方法学验证的阿魏酸钠标准曲线
附图5:实施例5方法学验证的桂利嗪标准曲线
附图6:实施例5方法学验证的阿司匹林标准曲线
具体实施例
实施例1
样品预处理筛选
(1)实验组A:参照蓝献泉等公开的《HPLC法同时测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、桂利嗪、阿司匹林、维生素B1》“溶液制备”部分的2.2.1供试品溶液、2.2.2混合对照品溶液、2.2.3阴性供试品溶液方法制备。
色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul;。
(2)实验组B:本发明方法。
色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
供试品溶液制备:取复方阿魏酸钠阿司匹林胶囊20粒内容物置研砵,研细,精密称取约相当于阿魏酸钠50mg、阿司匹林20mg、桂利嗪25mg的细粉(约相当于一粒的量)置100ml量瓶中,加1%冰醋酸甲醇溶液适量,超声(40kHz,250W)振荡2分钟使溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
混合对照品溶液制备:精密称取阿魏酸钠对照品约50mg,阿司匹林对照品约20mg,桂利嗪对照品约25mg同置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为对照品溶液。
阴性对照品溶液制备:取混合辅料空白,精密称取约1粒的量,同法制备,作为阴性对照品溶液。
测定方法:分别吸取供试品溶液、混合对照品溶液、阴性对照品溶液各10ul,注入液相色谱仪,测定,记录色谱图,以峰面积按外标法计算各组分标示百分含量,即得。
(3)实验组C:国家药品标准(新药转正标准)第45册的UV法测定阿魏酸钠和阿司匹林含量,重量法测定桂利嗪含量,不测定维生素B1含量。
实验结果:(1)实验组A使用流动相甲醇-乙腈-醋酸水(25∶20∶55)溶解样品超声过程中,样品的包衣聚团,影响了样品中各组分溶解程度。实验组B样品的溶解使用1%冰醋酸甲醇溶液配制简单快速,无需调节PH值,溶解样品快速,2分钟即可把样品中的阿魏酸钠、阿司匹林、桂利嗪溶解完全,溶解过程中没有包衣聚团。
(2)实验组B中供试品溶液的制备和对照品溶液的制备均一步到位,而附件中的方法(韶关药检所方法)供试品溶液制备需要两步稀释,对照品溶液的制备需要3步稀释,操作复杂,易由于多次移液转移定容产生操作误差。
(3)实验组A测定的阿司匹林、桂利嗪、阿魏酸钠和维生素B1含量比实验组B、实验组C和生产理论值低。而实验组A阿司匹林比其他组低20%左右,实验组A桂利嗪比其他组也是20%左右,分析原因可能是实验组A样品溶解过程中形成包衣聚团,导致样品中各组分的溶解受到影响,而阿司匹林和桂利嗪溶解性比较差,导致阿司匹林和桂利嗪难以溶解;并且实验组A使用溶解样品的初始比例流动相中含有55%水相且超声长达15分钟,导致样品中的阿司匹林快速水解产生大量的水杨酸,这些原因导致阿司匹林损失严重。
(4)实验组A测定的阿司匹林、桂利嗪含量比实验组B、实验组C和生产理论值低,分析原因可能是实验组A样品溶解过程中形成包衣聚团,导致样品中的桂利嗪难以溶解;溶剂中含水相较多,样品中的阿司匹林在超声15min过程中迅速水解致含量偏低。
结论:本发明所述样品预处理方法优于现有技术。
表格1:各实验组测定阿司匹林、桂利嗪、阿魏酸钠和维生素B1的标示百分含量(%)
实施例2色谱条件筛选
(1)实验组D:参照蓝献泉等公开的《HPLC法同时测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、桂利嗪、阿司匹林、维生素B1》优选出的最佳实验条件操作。
(2)实验组E:本发明所述方法。色谱柱:以十八烷基硅烷键合硅胶为填充剂;流动相:以乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70)为流动相A,甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4)用3mol/L盐酸溶液调节PH=6.6为流动相B,按下表2进行梯度洗脱;检测波长为276nm,以以下表格2中梯度进行试验。
本实施例中实验溶液的配制方法为:精密称取阿魏酸钠对照品约50mg,阿司匹林对照品约20mg,桂利嗪对照品约25mg同置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为对照品溶液。
表2流动相梯度
结论:表3、表4、图1、图2为实验组D和实验组E的实验结果,从2个表格和附图中可以看出实验组D中桂利嗪其峰型不对称,拖尾因子超1.5,而本发明方法中各成分峰的分离度良好,保留时间适中,各峰理论塔板数高,峰型对称。
表3实验组D各组分系统适用性结果
表4实验组E各组分系统适用性结果
实施例3:流动相梯度筛选
色谱柱:以十八烷基硅烷键合硅胶为填充剂;流动相:以乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70)为流动相A,甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4)用3mol/L盐酸溶液调节PH=6.6为流动相B,按下表进行梯度洗脱;检测波长为276nm,以以下表5梯度进行试验。比较流动相的不同梯度时间变化,以优化更佳的色谱条件。
本实施例中实验溶液的配制方法为:精密称取阿魏酸钠对照品约50mg,阿司匹林对照品约20mg,桂利嗪对照品约25mg同置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为对照品溶液。
表5梯度优化结果比较
结论:最终选定色谱条件为:以十八烷基硅烷键合硅胶为填充剂;流动相:以乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70)为流动相A,甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4)用3mol/L盐酸溶液调节PH=6.6为流动相B,按上表序号3方法进行梯度洗脱;检测波长为276nm为色谱条件。
实施例4:
(1)色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
(2)供试品溶液制备:取复方阿魏酸钠阿司匹林胶囊20粒内容物置研砵,研细,精密称取约相当于阿魏酸钠50mg、阿司匹林20mg、桂利嗪25mg的细粉(约相当于一粒的量)置100ml量瓶中,加1%冰醋酸甲醇溶液适量,超声(40kHz,250W)振荡2分钟使溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
(3)混合对照品溶液制备:精密称取阿魏酸钠对照品约50mg,阿司匹林对照品约20mg,桂利嗪对照品约25mg同置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为对照品溶液。
(4)阴性对照品溶液制备:取混合辅料空白,精密称取约1粒的量,同法制备,作为阴性对照品溶液。
(5)测定方法:分别吸取供试品溶液、混合对照品溶液、阴性对照品溶液各10ul,注入液相色谱仪,测定,记录色谱图,以峰面积按外标法计算各组分标示百分含量,即得。
实施例5:方法学验证
(1)专属性试验
阴性对照品溶液制备:取混合辅料空白,精密称取0.277g(约1粒的量),置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为阴性对照品溶液。
阿魏酸钠工作对照品溶液制备:精密称取阿魏酸钠对照品约50mg,置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为阿魏酸钠工作对照品溶液。
阿司匹林对照品溶液制备:临用新配,精密称取阿司匹林对照品约20mg,置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为阿司匹林对照品溶液。
桂利嗪工作对照品溶液制备:精密称取桂利嗪工作对照品约25mg,置100ml量瓶中,加1%冰醋酸甲醇溶液超声2分钟使溶解并稀释至刻度,摇匀,作为桂利嗪工作对照品对照品溶液。
色谱条件:本发明所述方法。
实验结果见附图3,结果显示,辅料空白样品图谱中在阿魏酸钠峰、阿司匹林峰、桂利嗪峰相同的保留时间位置处未出峰,说明辅料空白无干扰,该方法的专属性较强。
(2)稳定性
混合对照品溶液制备:精密称取阿魏酸钠工作对照品54.30mg、阿司匹林对照品20.34mg和桂利嗪工作对照品25.02mg置100ml量瓶中,加1%冰醋酸甲醇溶液溶解,并稀释至刻度,摇匀,备用。于室温下放置24小时,分别于0、2、4、6、8、24小时精密量取10ul按上述含量测定方法测定,结果见表6。
色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
表6溶液稳定性试验结果
结论:试验结果显示,溶液中阿魏酸钠稳定性RSD为1.3%;桂利嗪稳定性RSD为1.4%;阿司匹林稳定性RSD为9.0%(超出目标值),表明溶液中阿魏酸钠、桂利嗪在24h内稳定,阿司匹林在溶液中会逐渐产生游离水杨酸,经计算4、6、8小时内的RSD分别为1.3%、2.9%、3.9%,表明溶液中阿司匹林在4小时内较稳定,但为试验结果更准确,因此要求试验中样品溶液必须临用新配。
(3)回收率试验
分别精密称取理论检测浓度的80%、100%、120%的阿魏酸钠对照品、阿司匹林对照品、桂利嗪对照品即含阿魏酸钠约40、50、60mg、阿司匹林约16、20、24mg、桂利嗪约20、25、30mg的各3份置100ml量瓶中,再分别精密称取约0.277g理论处方量的混合辅料,加1%冰醋酸甲醇溶液溶解并稀释至刻度,摇匀,滤过作为供试品溶液。
色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
计算回收率,回收率相关数据及结果见表7、表8、表9和表10。
表7准确度试验称样量
表8阿魏酸钠准确度试验结果
表9阿司匹林准确度试验结果
表10桂利嗪准确度试验结果
结论:结果表明:阿魏酸钠、阿司匹林、桂利嗪三组分的回收率均在95%~105%,平均回收率分别为101.0%、101.5%、100.1%表明该方法准确度良好。
(4)精密度试验
混合对照品溶液制备:精密称取阿魏酸钠工作对照品54.30mg、阿司匹林对照品20.34mg和桂利嗪工作对照品25.02mg置100ml量瓶中,加1%冰醋酸甲醇溶液溶解,并稀释至刻度,摇匀,作为混合对照品溶液。
色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%;流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
精密吸取对照品溶液10ul,连续进样6次,测定,计算其峰面积RSD,结果见表11。
表11精密度试验结果
结论:试验结果表明,对照品溶液中阿魏酸钠精密度RSD为1.1%;桂利嗪稳定性RSD为1.2%;阿司匹林稳定性RSD为1.1%,表明该方法进样精密度良好。
(5)重复性试验
供试品溶液制备:临用新配,取复方阿魏酸钠阿司匹林胶囊20粒内容物置研砵,研细,精密称取约相当于阿魏酸钠50mg、阿司匹林20mg、桂利嗪25mg及10mg维生素B1的细粉(约相当于一粒的量)置100ml量瓶中,加1%冰醋酸甲醇溶液适量,超声2分钟使溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
色谱条件:以十八烷基硅烷键合硅胶为填充剂(推荐:月旭Xtimate C184.8×250mm,5um);流动相:A相为乙腈-四氢呋喃-冰醋酸-水(20∶5∶5∶70),B相为PH=6.6的甲醇-0.2%三乙醇胺-三乙胺(80∶20∶0.4),A相和B相梯度洗脱方法为:0-12.00min,A相100%;12.01-25.00min,B相100%;25.01-35.00min,A相100%:流速:1.0mL/min;检测波长:276nm;柱温:30℃;进样量:10ul。
取同一批样品(批号:140901),按照供试品溶液制备方法平行处理6份,结果如下表12:
表12重复性试验
结论:试验结果表明,6份样品含量测定结果RSD分别阿魏酸钠为0.3%;桂利嗪RSD为0.7%;阿司匹林RSD为0.4%,表明该方法重复性良好。
(6)线性试验
精密称取阿魏酸钠对照品约100.62mg(批号:1407020871)、阿司匹林对照品约40.39mg(批号:100113-201104)、桂利嗪对照品50.15mg(批号:150203-1)置100ml量瓶中,加1%冰醋酸甲醇溶液溶解并稀释至刻度,摇匀,作为对照品贮备溶液。用上述溶液,以1%冰醋酸甲醇作溶剂,按下表进一步稀释制成一系列线性测试溶液,浓度相当于对照品溶液浓度的40%-200%。取各溶液10ul注入液相色谱仪,按上述色谱条件检测,以峰面积为横坐标(X),对照品浓度(mg/ml)为(Y),绘制标准曲线。
表13线性试验
计算各组分回归方程
阿魏酸钠回归方程为:Y=0.0000591907X-0.011920712,r=0.9999;阿司匹林回归方程为:Y=0.000289771X-0.00907672,r=0.9999;桂利嗪回归方程为:Y=0.00019103X-0.004825443,r=0.9999,结果表明:阿魏酸钠在0.20124~1.0062mg/ml;阿司匹林在0.08278~0.4139mg/ml;桂利嗪在0.1003~0.5015mg/ml范围内呈良好的线性关系。
结论:方法学验证总评:以上各项目验证结果均符合中国药典相关规定,该方法准确、有效可行。
实施例6
在含量测定新方法建立后我们选定了三批样品与现行方法含量测定结果比较,结果见表14。
现行方法:国家药品标准WS1-(X-372)-2003Z(国家药品标准新药转正标准第45册45-203~45-205页)
新方法:本发明实施例4所述方法。
表14新方法与现行方法样品测定结果
结论:以上多批次样品的结果比较表明,新方法含量测定能够有效、准确的测定复方阿魏酸钠阿司匹林胶囊中阿魏酸钠、阿司匹林、桂利嗪的含量,方法稳定,可靠、高效,可定入复方阿魏酸钠阿司匹林胶囊质量标准中。
- 还没有人留言评论。精彩留言会获得点赞!