一种测定羧基麦芽糖铁分子量及其分布的方法与流程
本发明属于药物分析
技术领域:
,具体而言,涉及一种测定羧基麦芽糖铁分子量及其分布的方法。
背景技术:
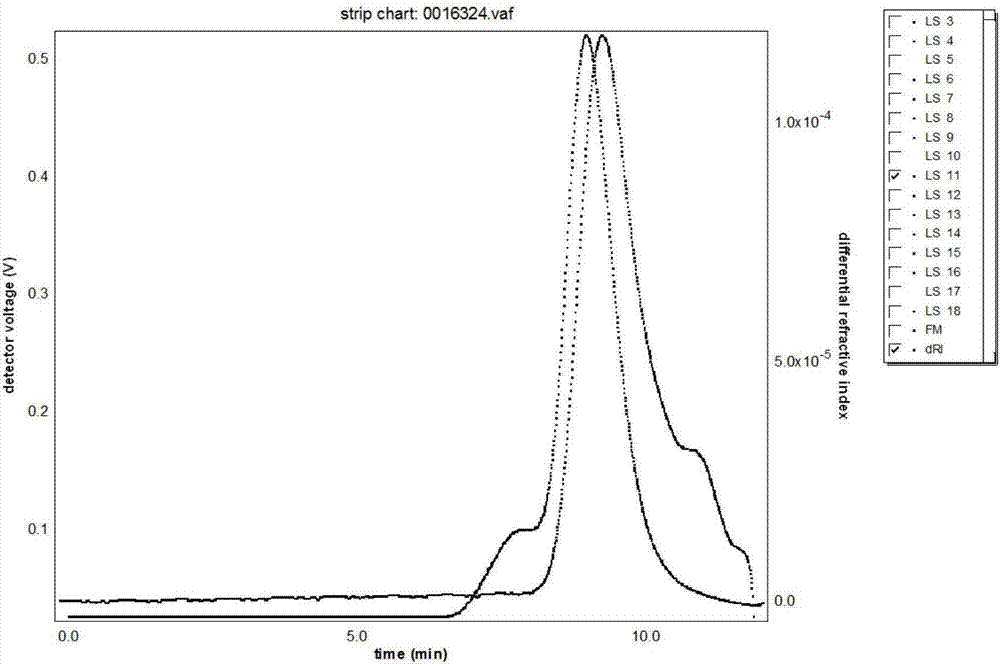
:缺铁性贫血至今仍是全球普遍而重要的健康问题,尤其是发展中国家。缺铁性贫血的治疗方法一般采用补铁药物治疗,目的在于补充血液和组织所缺的铁,使血红蛋白恢复正常水平,并补足贮存铁。口服补铁剂有效、价廉且安全,是缺铁性贫血的首选药物。但口服铁剂含铁率低,生物利用度低,常有明显的胃肠道不适症状,还受食物成份的干扰以及体内铁储备的影响,从而影响铁的吸收和利用,此时口服铁剂效果往往很差或根本无效。尤其是在许多慢性疾病(包括慢性心功能衰竭、肾功能衰竭等)和肿瘤患者中,体内炎性细胞介质增多,炎症介质(如白细胞介素-6等)能诱导肝脏合成铁调节蛋白,后者下调胃肠道及体内贮铁细胞表面膜铁转运蛋白的表达,从而影响铁的吸收和利用。此时,注射铁剂成为首选药物,它可更快地恢复铁储存,升高血红蛋白水平。目前世界上供临床应用的注射铁剂包括右旋糖酐铁、复合葡萄糖酸钠铁、蔗糖铁、山梨醇铁、异麦芽糖酐铁1000、ferumoxytol(多聚糖超顺磁氧化铁)和羧基麦芽糖铁。其中羧基麦芽糖铁由于具有良好的降解速率和物理化学特性,可以非常有效地将铁转运至体内贮存和转运蛋白,是一种比较理想的注射铁剂。羧基麦芽糖铁可以安全快速补铁并可替代输血,其克服了口服铁制疗程长、显效慢、费用高、胃肠道反应大、一天多次用药耐受性差的缺点,即使是消化道疾病引起的贫血也可使用;与低分子量的复合物如山梨醇铁相比,本品更有效,且毒性更小;本品不受与蔗糖铁和复合葡萄糖酸钠铁相同的高ph值和渗透压限制,而导致给药量受限,此外,其他静脉铁剂如蔗糖铁由于ph值和渗透压限制每次需要输注长达3.5小时(500mg铁);从铁转运和复合物稳定性来看,本品与右旋糖酐具有可比性,但其大大降低了右旋糖酐在临床应用中发生抗体形成或过敏反应的风险。此外,本品也克服了输血可能引发艾滋病、肝炎等感染风险和溶血反应,以及费用也居高不下的缺点。羧基麦芽糖铁由瑞士vifor公司研发,于2007年首先在德国上市,商品名ferinject。羧基麦芽糖铁是一种与羧基麦芽糖复合的胶体铁(iii)氢氧化物,是一种释放铁的糖聚物,其化学名称为多核铁(iii)氢氧化物4(r)-[聚-(1→4)-o-α-d-吡喃葡萄糖基]-氧基-2(r),3(s),5(r),6-四羟基己酸复合物,分子式:[feox(oh)y(h2o)z]n[{(c6h10o5)m(c6h12o7)}l]k,其中n≈103,m≈8,l≈11,k≈4(l代表配体的平均支化度),相对分子量:约为150,000da。羧基麦芽糖铁在制备过程中,由于反应参数控制的不同,其分子量以及分布会有很大的不同。目前,测定高聚物多糖分子量的方法所采用的标准物质往往与被测物质的化学结构不相同,再有标准物质分子量范围能否全覆盖被测样品分子量范围等问题。由此建立的为相对标准曲线,所测结果为相对分子量。受到上述条件的影响,所测的结果与物质的实际分子量有较大的误差。因此,需要建立一种适合工业生产的羧基麦芽糖铁分子量极其分布的测定方法。gpc-las联用技术兼具了gpc法和激光光散射法的特点,不需要采用标准物质做出标准曲线而直接快速、准确的测得羧基麦芽糖铁的重均分子量及分子量分布。技术实现要素:本发明的目的是提供一种不需要采用标准物质做出标准曲线而直接快速、准确的测得羧基麦芽糖铁的重均分子量及分子量分布的方法。为了达到上述目的,本发明提供了一种羧基麦芽糖铁分子量及分子量分布测定方法,其特征在于,具体步骤包括:将待检测的羧基麦芽糖铁用纯水溶解后,加入流动相,调节ph值到5-7,过滤,利用gpc系统进行分离的同时使用示差折光检测器和激光光散射检测器进行检测,将激光光散射检测器检测得到的分子量数据和示差折光检测器检测得到的浓度数据通过gpc软件计算得出羧基麦芽糖铁的分子量及分子量分布。优选地,进行分离和检测时,采用的流动相为超纯水、醋酸钠盐缓冲溶液、硝酸钠溶液和磷酸钠盐溶液中的至少一种,分析柱为1-4根分离范围为2,000,000~1000道尔顿的尺寸排阻色谱柱,柱温保持在20℃-40℃,流速为0.2ml/min—1.0ml/min。更优选地,进行分离和检测时,采用的流动相为ph=5.0~7.0的0.15m醋酸钠盐缓冲液。更优选地,进行分离和检测时,采用的分析柱为二根分离范围为1,000,000—1000道尔顿的亲水性凝胶柱串联。更优选地,进行分离和检测时,采用的柱温为35℃。更优选地,进行分离和检测时,采用的流速保持在0.8ml/min。更优选地,所述的gpc系统、示差折光检测器和激光光散射检测器的仪器常数校正时采用的标准物质为peg,验证仪器常数用的标准物质为葡聚糖右旋糖酐。与现有技术相比,本发明的有益效果是:本发明利用示差折光检测仪与光散射检测仪相连接(gpc-las法)测定羧基麦芽糖铁分子量及其分布,激光光散射检测器提供分子量的直接测量(无需校准曲线);使用示差折光检测器采集测定浓度,双检测器采集的信息通过gpc软件计算出分子量及其分布信息。本发明能够快速、准确的获得羧基麦芽糖铁的分子量及分子量分布,对提高该药品的内在品质具有积极意义。本发明发现羧基麦芽糖铁在水中易溶,其分离用gpc柱为亲水性球型高聚物为填充剂,该物质的ph耐受范围为3~11,通常在中性条件下使用。附图说明图1为柱温35℃时的gpc-las图谱;图2为流速0.8ml/min时的gpc-las图谱;具体实施方式以下通过实施例形式,对本发明涉及的羧基麦芽糖铁分子量以及分布测定方法作进一步的详细说明,但不应将此理解为本发明上述主题的范围仅限于以下的实例,凡基于本发明上述内容所实现的技术均属于本发明的范围。实施例1(1)仪器:高效凝胶渗透色谱仪为英国马尔文公司生产的omini工作站系统,optilabrex示差折光检测器,waters公司生产;515hplc泵,waters公司生产;270dual激光光散射检测器,英国马尔文公司生产;ve7510gpc脱气机,英国马尔文公司生产。其他具有上述配件及功能的高效液相色谱仪器亦可。(2)色谱条件:色谱柱:色谱柱为shodexohpaksb-806mhq凝胶柱,日本shodex公司生产;流动相:以0.15mph为6.5的醋酸钠盐缓冲溶液为流动相。流速:0.8ml/min。进样量:200μl。精密称取待检测的羧基麦芽糖铁供试品100mg,置于10ml容量瓶中,精密量取纯水2.0ml加入容量瓶,溶胀并振摇溶液60min,使溶解;加流动相3ml,摇匀,精密量取0.5mol/l的氢氧化钠溶液2.0ml加入容量瓶使ph调节约6.5,再加流动相至容量瓶刻度,摇匀,过0.22um尼龙滤膜滤,即得供试品溶液。吸取供试品溶液200μl,注入液相色谱仪,利用gpc系统进行分离的同时使用示差折光检测器和激光光散射检测器进行检测,gpc软件工作站采集示差折光检测器和激光光散射检测器信号,并处理得到图谱。先后检测得到柱温为25℃,35℃,45℃时的gpc图谱,其中图形以35℃较好,见图1。实施例2(1)仪器:高效凝胶渗透色谱仪为英国马尔文公司生产的omini工作站系统,optilabrex示差折光检测器,waters公司生产;515hplc泵,waters公司生产;270dual激光光散射检测器,英国马尔文公司生产;ve7510gpc脱气机,英国马尔文公司生产。其他具有上述配件及功能的高效液相色谱仪器亦可。(2)色谱条件:色谱柱:色谱柱为shodexohpaksb-806mhq凝胶柱,日本shodex公司生产;流动相:以0.15mph为6.5的醋酸钠盐缓冲溶液为流动相。柱温:35℃。进样量:200μl。精密称取待检测的羧基麦芽糖铁供试品100mg,置于10ml容量瓶中,精密量取纯水2.0ml加入容量瓶,溶胀并振摇溶液60min,使溶解;加流动相3ml,摇匀,精密量取0.5mol/l的氢氧化钠溶液2.0ml加入容量瓶使ph调节约6.5,再加流动相至容量瓶刻度,摇匀,过0.22um尼龙滤膜滤,即得供试品溶液。吸取供试品溶液200μl,注入液相色谱仪,利用gpc系统进行分离的同时使用示差折光检测器和激光光散射检测器进行检测,gpc软件工作站采集示差折光检测器和激光光散射检测器信号,并处理得到图谱。先后检测得到流速为0.5ml/min,0.8ml/min,1.0ml/min时的gpc图谱,图形以0.8ml/min为优,见图2。实施例3(1)仪器:高效凝胶渗透色谱仪为英国马尔文公司生产的omini工作站系统,optilabrex示差折光检测器,waters公司生产;515hplc泵,waters公司生产;270dual激光光散射检测器,英国马尔文公司生产;ve7510gpc脱气机,英国马尔文公司生产。其他具有上述配件及功能的高效液相色谱仪器亦可。(2)色谱条件:色谱柱:选用二根分离范围为1,000,000~1000道尔顿的不锈钢尺寸排阻色谱柱串联,所述的色谱柱为shodexohpaksb-806mhq凝胶柱,日本shodex公司生产;流动相:以0.15mph为6.5的醋酸钠盐缓冲溶液为流动相。柱温:35℃;流速:0.8ml/min。进样量:200μl。(3)实验步骤:步骤a:精密称取已知分子量的peg(mw150k)20mg加流动相稀释,并搅拌45min,使其溶解制成浓度为10mg/ml的peg溶液,另取葡聚糖(mw200k)和右旋糖酐((mw100k)),同法制成浓度为10mg/ml的葡聚糖溶液。步骤b:取peg溶液200μl,注入液相色谱仪,记录色谱图由gpc软件处理计算。用peg溶液获得的数据校正仪器并建立方法,取葡聚糖溶液和右旋糖酐溶液200μl,注入液相色谱仪,再用葡聚糖溶液和右旋糖酐溶液的数据结果复核peg溶液数据校正仪器的准确性,其结果应在150k±5.0%。步骤c:精密称取待检测的羧基麦芽糖铁供试品100mg,置于10ml容量瓶中,精密量取纯水2.0ml加入容量瓶,溶胀并振摇溶液60min,使溶解;加流动相3ml,摇匀,精密量取0.5mol/l的氢氧化钠溶液2.0ml加入容量瓶使ph调节约6.5,再加流动相至容量瓶刻度,摇匀,过0.22um尼龙滤膜滤,即得供试品溶液。步骤d:吸取供试品溶液200μl,注入液相色谱仪,加入到gpc系统中,利用gpc系统进行分离的同时使用示差折光检测器和激光光散射检测器进行检测,将激光光散射检测器检测得到的分子量数据和示差折光检测器检测得到的浓度数据通过gpc软件计算得出羧基麦芽糖铁的分子量及分子量分布;为考察gpc-las系统的重现性,将一批羧基麦芽糖铁重复进样5次。其结果见表1。表11批羧基麦芽糖铁重复5次的gpc—las结果比较实施例4(1)仪器:高效凝胶渗透色谱仪为英国马尔文公司生产的omini工作站系统,optilabrex示差折光检测器,waters公司生产;515hplc泵,waters公司生产;270dual激光光散射检测器,英国马尔文公司生产;ve7510gpc脱气机,英国马尔文公司生产。其他具有上述配件及功能的高效液相色谱仪器亦可。(2)色谱条件:色谱柱:选用二根分离范围为1,000,000~1000道尔顿的不锈钢尺寸排阻色谱柱串联,所述的色谱柱为shodexohpaksb-806mhq凝胶柱,日本shodex公司生产;流动相:以0.15mph为6.5的醋酸钠盐缓冲溶液为流动相。柱温:35℃;流速:0.8ml/min。进样量:200μl。(3)实验步骤:步骤a:精密称取已知分子量的peg(mw150k)20mg加流动相稀释,并搅拌45min,使其溶解制成浓度为10mg/ml的peg溶液,另取葡聚糖(mw200k)和右旋糖酐((mw100k)),同法制成浓度为10mg/ml的葡聚糖溶液。步骤b:取peg溶液200μl,注入液相色谱仪,记录色谱图由gpc软件处理计算。用peg溶液获得的数据校正仪器并建立方法,取葡聚糖溶液和右旋糖酐溶液200μl,注入液相色谱仪,再用葡聚糖溶液和右旋糖酐溶液的数据结果复核peg溶液数据校正仪器的准确性,其结果应在150k±5.0%。步骤c:精密称取待检测的羧基麦芽糖铁供试品100mg,置于10ml容量瓶中,精密量取纯水2.0ml加入容量瓶,溶胀并振摇溶液60min,使溶解;加流动相3ml,摇匀,精密量取0.5mol/l的氢氧化钠溶液2.0ml加入容量瓶使ph调节约6.5,再加流动相至容量瓶刻度,摇匀,过0.22um尼龙滤膜滤,即得供试品溶液。步骤d:吸取供试品溶液200μl,注入液相色谱仪,加入到gpc系统中,利用gpc系统进行分离的同时使用示差折光检测器和激光光散射检测器进行检测,将激光光散射检测器检测得到的分子量数据和示差折光检测器检测得到的浓度数据通过gpc软件计算得出羧基麦芽糖铁的分子量及分子量分布;为考察gpc-las系统的重现性,测定三份不同批次(批号)羧基麦芽糖铁的gpc—las图谱,数据见表2。表2:3批羧基麦芽糖铁分子量测定结果批次1#2#3#mw(道尔顿)161185167936205863rsd(%)4.82.93.6实施例5几种分子量测定方法比较如下:一、本发明方法(gpc-las法):仪器和色谱条件与实施例3相同,测定结果见表3;二、传统gpc方法采用示差折光检测器:取重均分子量(mw)分别为1.0、5.0、20.0、40.0、80.0、100.0万道尔顿的葡聚糖各50mg加流动相溶解,得到浓度为10mg/ml的溶液,分别注入gpc系统,色谱条件与本发明方法相同,数据处理绘制成分子量标准曲线。精密称定重均分子量(mw)20.0万道尔顿的右旋糖酐50mg分别溶解,得到浓度为10mg/ml的右旋糖苷溶液;将批号1#的羧基麦芽糖铁样品作为供试品,精密称取待检测的羧基麦芽糖铁供试品100mg,置于10ml容量瓶中,精密量取纯水2.0ml加入容量瓶,溶胀并振摇溶液60min,使溶解;加流动相3ml,摇匀,精密量取0.5mol/l的氢氧化钠溶液2.0ml加入容量瓶使ph调节约6.5,再加流动相至容量瓶刻度,摇匀,过0.22um尼龙滤膜滤,即得供试品溶液。测定结果见表3;三、传统gpc方法采用示差折光检测器:取重均分子量(mw)分别为1.0、5.0、20.0、40.0、80.0、100.0万道尔顿的右旋糖酐各50mg加流动相溶解,得到浓度为10mg/ml的溶液,分别注入gpc系统,色谱条件与本发明方法相同,数据处理绘制成分子量标准曲线。精密称定重均分子量(mw)20.0万道尔顿的葡聚糖50mg分别溶解,得到浓度为10mg/ml的葡聚糖溶液;,将批号1#的羧基麦芽糖铁样品作为供试品,精密称取待检测的羧基麦芽糖铁供试品100mg,置于10ml容量瓶中,精密量取纯水2.0ml加入容量瓶,溶胀并振摇溶液60min,使溶解;加流动相3ml,摇匀,精密量取0.5mol/l的氢氧化钠溶液2.0ml加入容量瓶使ph调节约6.5,再加流动相至容量瓶刻度,摇匀,过0.22um尼龙滤膜滤,即得供试品溶液。测定结果见表3;表3多种测定方法比较根据上述结果可以看出:本发明gpc—las法测定出的标准样品数值与其报告标定数值误差≤5.0%,满足gpc方法对分析检测精度的要求;传统gpc方法(葡聚糖校正曲线)在测定与自身相同的葡聚糖样品时,其测定数值与其报告标定数值误差≤5.0%,精度较高,但在测定右旋糖酐标准样品时结果与标准样品标定值误差大于20%;传统gpc方法(右旋糖酐校正曲线)存在同样的情况:在测定与自身相同的右旋糖酐样品时,其测定数值与其报告标定数值误差≤5.0%,精度较高,但在测定葡聚糖标准样品时结果与标准样品标定值误差大于25%:这些测定结果的偏差是由于这些不同的多糖对照样品与测定样品之间结构差异所造成的。高分子多糖均有较长的主链或侧链构成。由于对照品与样品的结果不同,造成的了在相同的色谱条件下,其在溶液中的主链或侧链舒展状态差异较大。同等分子量的情况下,由于溶液中舒展状态的不同造成了体积的不同。根据gpc分离原理--尺寸排阻,即样品溶液进入色谱柱后根据分子体积大小,分子量大物质的先分离流出,分子量小的后分离流出。分子量的大小确定是根据标准样品的保留时间或体积(分离时分子先后流出的时间或体积)绘制成得分子量标准曲线与检测样品来对比保留时间或体积来计算。葡聚糖与右旋糖酐两种多糖,在标定分子量相同的情况下,由于在溶液中的分子舒展状态差异,造成了体积的差异,继而在分离时产生了流出时间或体积的不同,从而造成测定结果较大的差异。总之,从上述的测定结果可以发现本发明的检测方法,克服了传统gpc的这些缺点,测定结果准确可靠。羧基麦芽糖铁在没有标准品做对照品的时候,本发明能快速、准确地测定其分子量。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1