一种基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N‑亚硝胺的提取及测定方法与流程
本发明涉及一种基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的提取及测定方法,属于烟草化学检验技术领域。
背景技术:
烟草特有N-亚硝胺是胺类化合物在酸性条件下与亚硝化试剂反应而生成的,是一类受到广泛关注的致癌物质,不仅是霍夫曼清单和美国食品药品监督管理局“烟草制品及烟气中有害及潜在有害物质名单”中的重要物质,也是国际癌症研究机构“无烟气烟草制品中28种有害物质”名单中的重要成分。因此,准确分析烟草及烟草制品中的烟草特有N-亚硝胺含量,对评价烟草及烟草制品、保障烟草制品消费安全具有重要意义。
最早建立的烟草或烟草制品中烟草特有N-亚硝胺的检测方法有两种:第一种方法是样品经水提取后用二氯甲烷萃取,有机相过氧化铝固相萃取柱净化,净化液浓缩后再经气相色谱-热能分析联用仪分离检测。但是上述方法均要进行液液萃取、固相萃取净化、净化液浓缩等操作,样品前处理步骤繁琐,不利于大量样品的快速分析。此外,热能分析仪虽是分析烟草特有N-亚硝胺的专属型检测器,但其仍存在前处理时间长、效率低等问题。第二种方法是用缓冲盐溶液提取烟草及烟草制品,将提取液过滤后进行液相色谱-串联质谱分析。该方法前处理操作简单,但是样品溶液进行仪器分析前没有经过净化处理,因此存在较强的基质干扰现象,加大了仪器维护频率。
申请公开号为CN102012407A的发明专利公开了一种烟草特有N-亚硝胺的检测方法,在该方案中将萃取液流经固相萃取小柱,然后采用甲酸水淋洗,最后用氨水/甲醇洗脱,得到待测液。该方案中的固相萃取填料为N-乙烯基吡咯烷酮-苯磺酸共聚物,有机聚合物填料在有机溶剂中会溶胀,会在一定程度上影响填料的富集和净化效果。另外,该方案中也没有对影响萃取效果的各种参数进行优化。
液相色谱-串联质谱以其强大的色谱分离能力、良好的选择性和灵敏度,在复杂样品痕量物质的分析中展现出强大的优势,在烟草特有N-亚硝胺的分析中有广泛的应用。
技术实现要素:
本发明的目的是提供一种基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的提取方法。
本发明的另一个目的是提供一种基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的测定方法。
为了实现以上目的,本发明所采用的技术方案是:
一种基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的提取方法,包括以下步骤:
1)将烟草或烟草制品加入浓度为5~100mmol/L的乙酸铵溶液进行提取,得粗提取液;
2)将步骤1)所得的粗提取液流过经活化处理后的HiCapt MCX固相萃取柱,完成上样;
3)对步骤2)中完成上样后的HiCapt MCX固相萃取柱进行淋洗;
4)用丙酮、丙酮氨水混合液、丙酮甲酸混合液中的任意一种进行洗脱,即得N-亚硝胺的提取液;所述丙酮氨水混合液中丙酮、氨水的体积比为97~99:3~1;丙酮甲酸混合液中丙酮、甲酸的体积比为97~99:3~1。
上述烟草或烟草制品中烟草特有N-亚硝胺包括NNN、NNK、NAT和NAB。NNN、NNK、NAT和NAB的结构如图1中所示。
上述步骤1)中提取操作为:将烟草或烟草制品加入浓度为5~100mmol/L的乙酸铵溶液中,进行超声处理,然后分离不溶物,得粗提取液。
上述每1g烟草或烟草制品所用乙酸铵溶液的用量为10~100mL。
上述超声处理,超声功率280~700W,频率20~40kHz,时间≥10min。超声时间优选为10~60min。超声时间进一步优选为30min。
上述分离不溶物采用过滤或离心的方式。
上述过滤采用水相滤膜。
上述水相滤膜的孔径为0.20~0.50μm。优选为0.22μm或0.45μm。
上述水相滤膜为聚醚砜膜。
上述离心,是将超声处理后的混合液静置,然后进行离心,最后取上层清液。
上述静置时间为3~10min。优选为5min。
上述离心转速5000~10000rpm,时间为3~10min。离心时间优选为5min。
上述HiCapt MCX固相萃取柱为疏水/离子交换混合保留机理固相萃取柱。
上述HiCapt MCX固相萃取柱购自于维泰克科技(武汉)有限公司。
上述HiCapt MCX固相萃取柱具有疏水和强离子交换作用。
上述HiCapt MCX固相萃取柱中HiCapt MCX固相萃取填料的用量选择时,每将6ml粗提取液完成上样时,相应使用50~1000mg的HiCapt MCX固相萃取填料。优选地,6ml粗提取液对应使用500mgHiCapt MCX固相萃取填料。
上述粗提取液在上样前的pH值调至3.0~11。优选为4~6。
上述调节pH值的调节剂为乙酸或氨水。优选地,当使用乙酸作为pH调节剂时,选用浓度为10%的乙酸溶液。
上述活化处理操作为:依次用丙酮、甲醇水溶液、水流经HiCapt MCX固相萃取柱。
上述活化为依次用丙酮、甲醇体积分数为5%的甲醇水溶液、水流经固相萃取柱。所述丙酮、甲醇水溶液、水的体积比为1:2:2。
上述步骤3)中用甲醇水溶液作为淋洗液对HiCapt MCX固相萃取柱进行淋洗,所述甲醇水溶液中甲醇的体积分数为5~20%。
上述步骤3)中淋洗后,在真空泵的负压作用下将HiCapt MCX固相萃取柱中的淋洗液抽干净。
上述步骤4)丙酮氨水混合液中丙酮与氨水的体积比优选为99:1。
上述步骤4)丙酮甲酸混合液中丙酮与甲酸的体积比优选为99:1。
上述氨水的质量分数为35%。
上述洗脱,洗脱液的用量为:每将6ml粗提取液上样,淋洗,干燥后使用的洗脱液体积≥0.5ml。优选为1~2ml。进一步优选为1ml。
一种基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的测定方法,包括以下步骤:
1.N-亚硝胺提取液的制备
1.1向烟草或烟草制品中依次加入同位素内标和浓度为5~100mmol/L的乙酸铵溶液进行提取,得到混合液;
1.2将步骤1.1所得的混合液流过经活化处理后的HiCapt MCX固相萃取柱,完成上样;
1.3对步骤1.2中完成上样后的HiCapt MCX固相萃取柱进行淋洗;
1.4用丙酮、丙酮氨水混合液、丙酮甲酸混合液中的任意一种进行洗脱,即得N-亚硝胺的提取液;
2)N-亚硝胺的测定
将步骤1.4中所得的提取液吹干,再用复溶溶液进行复溶得待测液,然后对待测液进行液相色谱-串联质谱测定;所述复溶溶液由甲酸铵溶液和甲酸乙腈溶液配制而成。
上述同位素内标包括NNN-d4、NNK-d4、NAT-d4、NAB-d4。
上述同位素内标的加入量为:每1mL的乙酸铵溶液中每种同位素内标的质量为10ng。
上述步骤1.1中所得混合液在上样前pH调至3.0~11.0。
上述步骤1.4中丙酮与氨水体积比优选为99:1。丙酮与甲酸的体积比优选为99:1。
上述洗脱,洗脱液的用量为:每将6ml混合液上样,淋洗,干燥后使用的洗脱液体积≥0.5ml。优选为1~2ml。进一步优选为1ml。
上述复溶溶液由0.01mol/L的甲酸铵溶液与甲酸体积分数为0.1%的甲酸乙腈溶液按照1~2:1~2的体积比配制而成。
上述复溶溶液的用量为:每使用6ml混合液,复溶溶液体积≥0.1ml。优选为0.1~1ml。进一步优选为0.2ml。
上述步骤2)中液相色谱-串联质谱测定包括系列浓度的混合标准溶液的配制,配制步骤为:
以四种烟草特有N-亚硝胺NNN、NNK、NAT和NAB的标准品为溶质,以NNN-d4、NNK-d4、NAT-d4和NAB-d4为内标,甲醇为溶剂,配制系列浓度的混合标准溶液。
上述测定方法中的提取操作参数与上述基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的提取方法参数相同。
本发明中基于疏水离子交换固相萃取的烟草特有N-亚硝胺的测定方法中标准曲线的制作步骤为:
以四种烟草或烟草制品中烟草特有N-亚硝胺NNN、NNK、NAT和NAB的标准品为溶质,以NNN-d4、NNK-d4、NAT-d4和NAB-d4为内标,甲醇为溶剂,配制系列浓度的混合标准溶液,将系列浓度的混合标准溶液进行液相色谱-串联质谱分析,以目标分析物峰面积和相应同位素峰面积之比对目标分析浓度做回归分析,即得各目标分析物的标准曲线。
上述液相色谱-串联质谱分析的条件为:色谱柱:Poroshell 120EC-C18;柱温:40℃;进样量:5μL;流动相A:0.01mol/L甲酸铵溶液,流动相B:0.1%甲酸乙腈溶液;流速:0.4mL/min;流动相梯度程序:t=0min,10%B,t=2.0min,40%B,t=4.0min,60%B,t=6.0min,90%B,t=9.0min,90%B,t=9.5min,10%B,t=15.0min,10%B;质谱条件:电喷雾电压5000V;雾化气压力50psi;辅助雾化气压力50psi;气帘气压力35psi;离子源温度450℃;进口电压8V;出口电压10V;去簇电压40V;驻留时间40ms;监测模式:多反应监测。
上述多反应监测中NNN、NNK、NAT和NAB的定量离子对依次为178.1>148.2、208.1>122.1、190.1>160.1和192.1>162.2,相应的碰撞电压依次为15、16、15和17eV;NNN、NNK、NAT和NAB的定性离子对依次为178.1>120.1、208.1>106.1、190.1>106.1和192.1>133.1,相应的碰撞电压依次为15、16、15和17eV;NNN-d4、NNK-d4、NAT-d4和NAB-d4的定量离子对依次为182.1>152.2、212.1>126.1、194.1>164.1和196.1>166.2,相应的碰撞电压依次为15、16、15和17eV。
上述色谱柱:Poroshell 120EC-C18 3.0mm×100mm,2.7μm。
本发明的有益效果:
本发明的基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的提取方法中采用了HiCapt MCX疏水/离子交换混合保留机理固相萃取柱,由于烟草特有N-亚硝胺本身具有疏水性能,再加上其结构中都含有吡啶环,HiCapt MCX固相萃取柱与烟草特有N-亚硝胺能产生疏水作用和强阳离子交换作用,从而使得HiCapt MCX固相萃取填料对样品提取液具有良好的净化效果,能有效去除杂质及干扰物,提高测定方法的准确度和精密度,方法的相对回收率介于92.2%~108.9%之间,日内及日间精密度分别小于4.6%和4.2%。
本发明中的基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的测定方法,通过采用HiCapt MCX固相萃取柱对目标分析物进行提取,对目标分析物进行疏水和阳离子交换,达到对目标分析物的纯化作用,然后用液相色谱-串联质谱法定性、定量分析烟草及烟草制品中烟草特N-亚硝胺,本发明的测定方法具有样品前处理操作简单、快捷,杂质及干扰物的去除效率高,处理时间短的优点。
附图说明
图1为混合标准溶液3的色谱图;
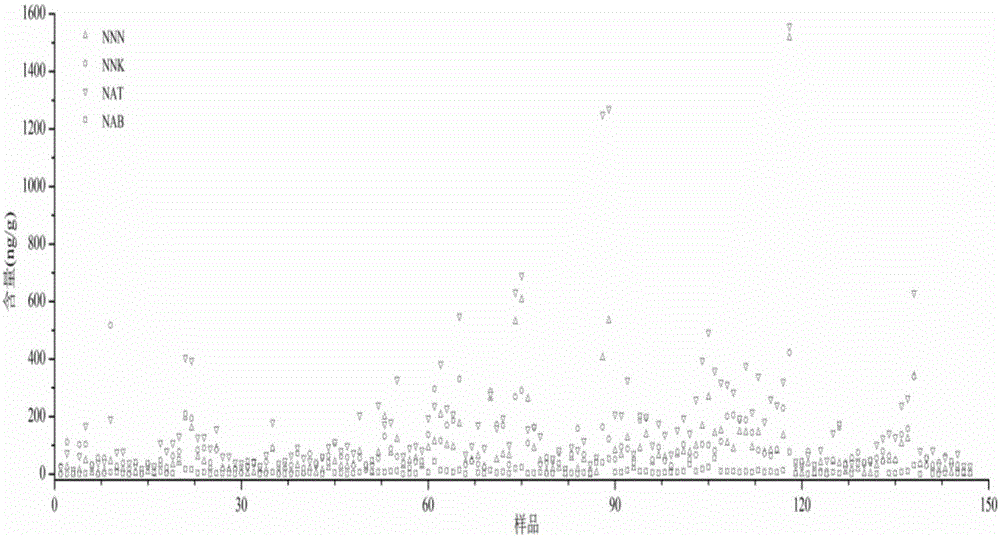
图2为上百种烟草或烟草制品的测定结果图;
图3为粗提取液pH值的优化结果图;
图4为洗脱液种类的优化结果图;
图5为洗脱液体积的优化结果图;
图6为淋洗液中甲醇含量的优化结果图;
图7为HiCapt MCX固相萃取柱中填料质量的优化结果图。
具体实施方式
下述实施例对本发明作进一步详细说明。
实施例1
本实施例中基于疏水离子交换固相萃取的烟草或烟草制品中烟草特有N-亚硝胺的提取方法,包括以下步骤:
1)称取1.0g烟草(精确至0.1mg),置入30mL具塞离心管中,准确加入30mL浓度10mmol/L的乙酸铵水溶液,置于超声波发生器(购自昆山市超声仪器有限公司,型号:KQ-700DB)上进行超声处理,超声提取30min,超声功率700W,频率40kHz,静置5min后,置于离心机上离心5min,离心转速10000rpm,取上层清液,即得粗提取液;粗提取液在上样前的pH值调至4,作为上样溶液;
2)准确称取500mg HiCapt MCX固相萃取填料于带有筛板的固相萃取空柱管中,不断敲打使填料填充均匀,上端盖上筛板压紧,待用;上样前,依次用1.0mL丙酮、2.0mL甲醇体积分数为5%的甲醇水溶液、2mL水活化固相萃取小柱;之后在重力作用下将6mL步骤1)中所得的上样溶液自然流过固相萃取小柱以完成上样过程;
3)上样结束后用1mL甲醇体积分数为10%的甲醇水淋洗液进行淋洗,待淋洗液完全流过萃取柱后,在真空泵的负压作用下将HiCapt MCX固相萃取柱中的淋洗液抽干净;
4)最后用2mL丙酮(含1%体积分数,质量分数为35%的氨水)洗脱目标分析物,收集,即得。
本实施例的基于疏水离子交换固相萃取的烟草中烟草特有N-亚硝胺的测定方法,包括以下步骤:
1.N-亚硝胺提取液的制备
1.1称取1.0g烟草(精确至0.1mg),置入30mL具塞离心管中,加入同位素内标后,准确加入30mL浓度10mmol/L的乙酸铵水溶液,置于超声波发生器(购自昆山市超声仪器有限公司,型号:KQ-700DB)上进行超声处理,超声提取30min,超声功率700W,频率40kHz,静置5min后,置于离心机上离心5min,离心转速10000rpm,取上层清液,即得粗提取液;粗提取液在上样前的pH值调至4,作为上样溶液;
1.2准确称取500mg HiCapt MCX固相萃取填料于带有筛板的固相萃取空柱管中,不断敲打使填料填充均匀,上端盖上筛板压紧,待用。上样前,依次用1.0mL丙酮、2.0mL甲醇体积分数为5%的甲醇水溶液、2mL水活化固相萃取小柱。之后在重力作用下将步骤1.1中所得的6mL上样溶液自然流过固相萃取小柱以完成上样过程;
1.3上样结束后用1mL甲醇体积分数为10%的甲醇水淋洗液进行淋洗,待淋洗液完全流过萃取柱后,在真空泵的负压作用下将HiCapt MCX固相萃取柱中的淋洗液抽干净;
1.4用2mL丙酮(含1%体积分数,质量分数为35%的氨水)洗脱目标分析物,收集,即得。
2.N-亚硝胺的测定
将步骤1.4中所得提取液在35℃下氮气吹干,再用0.2mL复溶溶液进行复溶。复溶溶液由0.01mol/L的甲酸铵溶液和甲酸体积分数为0.1%的甲酸乙腈混合溶液按照1:1的体积比配制而成。
混合标准溶液的配制:以四种烟草特有N-亚硝胺的同位素内标(NNN-d4、NNK-d4、NA-d4T和NAB-d4)为溶质,甲醇为溶剂,配制内标溶液;以四种烟草特有N-亚硝胺(NNN、NNK、NAT和NAB)为溶质,甲醇为溶剂,配制系列浓度的混合标准溶液,具体见下表1,其中内标的浓度均为10ng/mL。将混合标准溶液进行液相色谱-串联质谱分析。
表1混合标准溶液的浓度表
标准曲线的制作:取混合标准溶液进行液相色谱-串联质谱分析,分析条件为:色谱柱:Poroshell 120EC-C18(3.0mm×100mm,2.7μm);柱温:40℃;进样量:5μL;流动相为0.01mol/L甲酸铵(A)-0.1%甲酸乙腈(B);流速:0.4mL/min;流动相梯度程序:t=0min,10%B,t=2.0min,40%B,t=4.0min,60%B,t=6.0min,90%B,t=9.0min,90%B,t=9.5min,10%B,t=15.0min,10%B;质谱条件:电喷雾电压5000V;雾化气(Gas1)压力50psi;辅助雾化气(Gas2)压力50psi;气帘气(Gas1)压力35psi;离子源温度450℃;进口电压8V;出口电压10V;去簇电压40V;驻留时间40ms;监测模式:多反应监测。
所述多反应监测中NNN、NNK、NAT和NAB的定量离子对依次为178.1>148.2、208.1>122.1、190.1>160.1和192.1>162.2,相应的碰撞电压依次为15、16、15和17eV;NNN、NNK、NAT和NAB的定性离子对依次为178.1>120.1、208.1>106.1、190.1>106.1和192.1>133.1,相应的碰撞电压依次为15、16、15和17eV;NNN-d4、NNK-d4、NAT-d4和NAB-d4的定量离子对依次为182.1>152.2、212.1>126.1、194.1>164.1和196.1>166.2,相应的碰撞电压依次为15、16、15和17eV。程序运行结束,以目标分析物峰面积和相应同位素峰面积之比对目标分析物浓度作回归分析,即得各目标分析物的标准曲线,参数见下表2,混合标准溶液3的色谱图见图1。
表2各目标分析物对应的标准曲线、定量限和检测限
注:检测限和定量限是信噪比(S/N)3和10时对应的浓度
样品中烟草特有N-亚硝胺的测定:取上述待测溶液进行液相色谱-串联质谱分析,分析条件同上,将测得的各物质峰面积代入下式1中,经计算得到样品中目标分析物的含量。
式1:
式中,m为样品中目标分析物的含量,ng/g;x为目标分析物与同位素内标的峰面积之比;a为标准曲线的斜率,b为标准曲线的截距;V为样品提取液的体积,mL;n为样品质量,g。
实施例2
按实施例1所述的测定方法,选取另外上百种烟草或烟草制品,测定其中烟草特有N-亚硝胺的含量,结果如图2所示。
本发明中的其他实施例中,粗提取液的pH可以调至3.4-11.0,进一步的本发明中的其他实施例中粗提取液的pH可以选择为3.4、6、10.5或11,其它同实施例1。由于HiCapt MCX固相萃取柱中的填料对目标分析物的萃取主要是靠疏水和阳离子交换作用,pH在4.0-6.0时大部分目标分析物分子以质子化形式存在,而填料中的阳离子交换基团以负离子形式存在,此时填料和目标分析物之间的疏水作用和阳离子交换作用力较强,因此具有最佳的萃取效果。如图3所示,在所考察的pH值范围(3.4-11.0)内,在pH为4.0时,所有被分析物具有较好的萃取效果。
本发明中的其他实施例中,步骤4)及1.4中洗脱液可选用丙酮、丙酮氨水混合液、丙酮甲酸混合液中的任意一种。进一步的,还可以为含3%体积分数氨水的丙酮混合液、含1%或3%体积分数甲酸的丙酮混合液或丙酮,其它同实施例1。如图4、图5洗脱液种类及体积优化结果图所示,在丙酮中添加一定比例的氨水,能够提高目标分析物的解吸效率,这是因为碱性条件下能够破坏填料和目标分析物之间的阳离子交换作用。
本发明中的其他实施例中,步骤3)及1.3中淋洗所用淋洗液的甲醇含量可以为5~20%。进一步地,甲醇水溶液中甲醇的体积分数还可以为5%、8%、10%、12%、15%或20%,其它同实施例1。如图6淋洗液中甲醇含量的优化结果图所示,当甲醇含量为10%时,目标分析物具有较好的萃取效率。
本发明中的其他实施例中,HiCapt MCX固相萃取柱中的填料的质量可以在50-1000mg范围内。进一步地,HiCapt MCX固相萃取柱中的填料的用量还可以为50mg、100mg、200mg、500mg或1000mg,其它同实施例1。如图7所示,填料的质量在50-1000mg范围内,随着填料质量不断提高,目标分析物的峰面积均显著提高,当填料的质量达到500mg时,所有目标分析物的萃取效果达到最优。
在本发明其它的实施例中,超声提取、漩涡振荡的条件均可适当调整。
试验例
精密度及加标回收测试
为考察上述测定方法的抗干扰性,在实施例1的待测样品中加入一定量的混合标准溶液,配制成低、中、高三种浓度的样品(低、中、高浓度对应的值分别为各目标分析物线性范围最低值的2、10和40倍)。在优化的分析条件下,目标分析物与干扰物分离良好,干扰物不影响目标分析物的定量。
以一天内配制的5个样品进行测定,计算不同浓度下的日内相对标准偏差;以连续3天配制的样品进行测定,计算不同浓度下的日间相对标准偏差;将分析所得的峰面积之比代入标准曲线中,计算得到待测溶液中目标分析物的含量,并与实际添加量相比得到相对回收率,结果见下表3。从表3可以看出,目标分析物在不同浓度下的日内及日间精密度分别小于4.6%和4.2%,说明该方法具有较好的重现性。同时,不同浓度下目标分析物的相对回收率介于92.2%~108.9%之间。为了进一步验证本方法的准确性,采用本方法和现行标准方法(YQ/T29-2013)对烟草及烟草制品进行分析,结果如表4所示。结果表明,两种方法得到的测定结果一致性良好,说明该方法的准确性好,可以满足日常分析要求。
表3精密度及加标回收测试结果
表4采用本方法和标准方法对烟草及烟草制品分析的结果(ng/g)
- 还没有人留言评论。精彩留言会获得点赞!