一种蟾酥中化合物的检测方法与流程
本发明涉及一种蟾酥中化合物的检测方法。
背景技术
蟾酥为国家保护药材,为国家“三有”动物蟾蜍耳后腺白色分泌物的干燥物,具有解毒,止痛,开窍醒神之功。蟾酥主治痈疽疔疮,咽喉肿痛,中暑神昏,腹痛吐泻,以蟾酥入药的方剂共88种,其质量的好坏直接影响制剂药效。目前,蟾酥评价标准主要是以每种单项成分的含量指标来表征其质量,已被广泛使用的外标法在实验时需要多个标准品对药材中的多种成分进行对应分析,这种方法耗时较长,标准品成本高以及标准品的高纯度因素限制了其应用。因此,亟需一种简单、准确的方法来检测蟾酥中各化合物组分的含量。
技术实现要素:
本发明提供了一种缩短检测用时间、检测时间准确、检测成本低的蟾酥中化合物的检测方法,解决了现有技术中存在的问题。
本发明为解决上述技术问题所采用的技术方案是:一种蟾酥中化合物的检测方法,
一种蟾酥中化合物的检测方法,其特征在于:包括如下步骤:
(1)对照品溶液的制备:以甲醇为溶剂,分别以沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基、酯蟾毒配基六种对照品为溶质,制得六种单项对照品标准溶液,将适量六种单项对照品标准溶液混合后并加入适量甲醇制得混合对照品标准溶液;
(2)供试品溶液的制备:取蟾酥粉末样品,加入甲醇溶液,称定重量,加热回流提取,放置冷却至室温,再称定重量,用甲醇补充减失的重量,摇匀,滤过,取续滤液,即得;
(3)确定色谱条件:色谱柱:填充剂为十八烷基硅烷键合硅胶,柱温25℃,检测波长296nm,流速0.5-0.7mlmin-1,进样量为3-20μl,流动相为0.1%甲酸(a)-乙腈(b),梯度洗脱;
(4)确定相对校正因子:分别取步骤(1)制得的混合对照品溶液3μl,5μl,7μl,9μl,10μl,13μl,15μl,17μl,在所述不同进样体积下分别进样,进行色谱条件检测分析,得到液相色谱图并计算各色谱峰的峰面积,选定华蟾酥毒基为内标物k,根据公式fk/m=fk/fm=wk×am/(wm×ak),分别计算每个进样体积下华蟾酥毒基与其他组分m间的相对校正因子,取平均值;其中,m为沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、酯蟾毒配基中的一种,am、ak为组分峰面积;wm、wk为组分浓度;
(5)供试品溶液组分含量的测定:取步骤(2)制得的供试品溶液进行高效液相色谱分析,获得液相色谱图,根据相对保留时间进行峰定位,计算各色谱峰的峰面积,然后结合华蟾酥毒基为内标物得到的相对校正因子计算沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、酯蟾毒配基的含量,并与外标法测定的结果进行比较分析。
步骤(1)制得的每种单项对照品标准溶液中溶质的浓度分别为2mg/ml。
步骤(1)制得的混合对照品标准溶液中各溶质的浓度分别为:沙蟾毒精0.045mg/ml、远华蟾毒精0.009mg/ml、蟾毒它灵0.03mg/ml、蟾毒灵0.025mg/ml、华蟾酥毒基0.051mg/ml、酯蟾毒配基0.031mg/ml。
步骤(3)所述梯度洗脱条件为:0~20min,80%(a);20~40min,70~65%(a);40~60min,65~40%(a)。
本发明采用上述方法,不但解决了对照品昂贵稀少的问题,还依从了环境友好、绿色中药的原则,选择含量高、峰值明显的华蟾酥毒基为内标物,可同时测量蟾酥中六种化合物的含量,大大提高检测效率,更加全面控制蟾酥的质量。根据检测对象确定合理的流动相、色谱柱、洗脱程序、检测波长、柱温等色谱条件,在选定的色谱条件下,快速分析测得蟾酥中化合物的含量,精密度高、重现性好、稳定性好、回收率高、测定结果准确。
附图说明
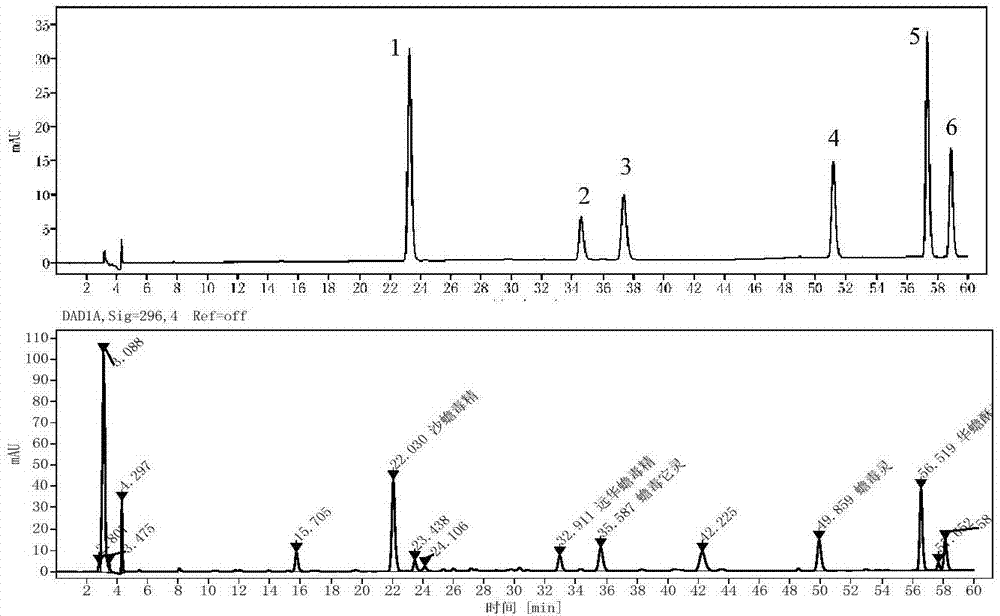
图1为本发明混合对照品溶液及供试品溶液的hplc图。
具体实施方式
为能清楚说明本方案的技术特点,下面通过具体实施方式,并结合其附图,对本发明进行详细阐述。
1仪器与试剂:
对照品:沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵(购于天津万象恒远科技有限公司)、华蟾酥毒基和酯蟾毒配基(购于中国食品药品检定研究院);
供试品:41份蟾酥样品;
仪器:agilent1260高效液相系统、waters2690高效液相系统;
试剂:乙腈(merck,色谱纯),甲酸(merck,色谱纯),纯水;
2方法与结果:
2.1对照品溶液的制备:
以甲醇为溶剂,精密称取沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基、酯蟾毒配基对照品各10mg为溶质,置于5ml量瓶中,制得浓度分别为2mg/ml的六种单项对照品标准溶液,精密吸取适量上述六种单项对照品标准溶液混合后,加入适量甲醇制得混合对照品标准溶液,混合对照品标准溶液中各溶质的浓度分别为:沙蟾毒精0.045mg/ml、远华蟾毒精0.009mg/ml、蟾毒它灵0.03mg/ml、蟾毒灵0.025mg/ml、华蟾酥毒基0.051mg/ml、酯蟾毒配基0.031mg/ml。
所用甲醇为纯甲醇。
2.2供试品溶液的制备:精密称定蟾酥粉末样品25mg,置于具塞锥形瓶中,加入甲醇溶液20ml,称定重量,在甲醇沸腾温度下加热回流提取1h,放置冷却至室温,再称定重量,用甲醇补充减失的重量,摇匀,滤过,取续滤液,即得;
2.3制定色谱条件:
色谱柱:agilentzorbaxsbc18(250mm×4.6mm,5μm);
流动相:0.1%甲酸(a)-乙腈(b),流速0.7mlmin-1,柱温25℃,检测波长296nm,流速0.7mlmin-1,进样量为3-20μl,流动相为0.1%甲酸(a)-乙腈(b),梯度洗脱序列见下表1,
表1流动相梯度洗脱程序
2.4确定相对校正因子:分别取步骤2.1制得的混合对照品溶液3μl,5μl,7μl,9μl,10μl,13μl,15μl,17μl,在所述不同进样体积下分别进样,进行色谱条件检测分析,得到液相色谱图并计算各色谱峰的峰面积(从图1中可看到混合对照品溶液中各化合物组分的分离度良好),选定华蟾酥毒基为内标物k,根据公式fk/m=fk/fm=wk×am/(wm×ak),分别计算每个进样体积下华蟾酥毒基与其他组分m间的相对校正因子,见表2,取平均值;其中,m为沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、酯蟾毒配基中的一种,am、ak为组分峰面积;wm、wk为组分浓度;
表2华蟾酥毒基对沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵和酯蟾毒配基的校正因子
2.4.1对照品的线性范围考察:
以化合物浓度对峰面积积分值进行回归处理,得沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基和酯蟾毒配基回归方程,分别为:y=13748x+236.06(r2=0.9999),y=9980.3x+34.599(r2=0.9996),y=8447.2x+96.422(r2=0.9998),y=11592x+114.78(r2=0.9990),y=15154x+286.40(r2=0.9998),y=8685.8x+103.36(r2=0.9998)。可见,六个化合物在0.010-0.557mg/ml,0.002-0.090mg/ml,0.005-0.501mg/ml,0.005-0.478mg/ml,0.008-0.914mg/ml,0.005-0.639mg/ml范围内线性良好。
2.5供试品溶液组分含量的测定:取步骤(2)制得的供试品溶液进行高效液相色谱分析,获得液相色谱图,根据相对保留时间进行峰定位,计算各色谱峰的峰面积,然后结合华蟾酥毒基为内标物得到的相对校正因子计算沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、酯蟾毒配基的含量,并与外标法测定的结果进行比较分析。
2.5.1精密度试验
精密吸取同一混合对照品溶液20μl,连续进样6次,记录峰面积,结果见表3。
2.5.2稳定性试验
取同一供试品溶液分别于2h,4h,6h,12h,24h,48h进行分析,记录峰面积,结果见表3。
2.5.3重复性试验
称取蟾酥药材粉末约25mg共6份,精密称定,按供试品溶液处理方法制备样品,测定,结果见表3。
表3沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基和酯蟾毒配基的精密度、重复性和稳定性
rrt为相对保留时间,rpa为相对峰面积
2.5.4加样回收率
精密称取适量蟾酥药材粉末6份,分别加入一定量的对照品,按供试品溶液处理方法制备样品,测定,计算加样回收率。结果见表4。
表4沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基和酯蟾毒配基的加样回收率
2.5.5样品测定
分别精密吸取供试品、混合标准品及单一标准品(华蟾酥毒基)溶液注入高效液相色谱仪,测定。分别采用一测多评法和外标法计算蟾酥药材中沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基和酯蟾毒配基的含量,见表5。
表5不同蟾酥样品中沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵、华蟾酥毒基和酯蟾毒
配基的含量
3本发明检测方法耐用性和系统适用性评价
3.1色谱柱及高效液相色谱仪考察
取2.1项下混合对照品溶液,进样测定,计算华蟾酥毒基对沙蟾毒精、远华蟾毒精、蟾毒它灵、蟾毒灵和酯蟾毒配基的校正因子。
考察了agilent1260和waters2695两种高效液相色谱系统和agilentzorbax-sbc18(250mm×4.6mm,5μm)、agilentzorbax-bonus-rp(250mm×4.6mm,5μm)和alltimac18(250mm×4.6mm,5μm)三种色谱柱,结果见表6。
表6.不同仪器和色谱柱的校正因子(n=6)
fc/a:华蟾酥毒基对沙蟾毒精;fc/t:华蟾酥毒基对远华蟾毒精;fc/bt:华蟾酥毒基对蟾毒它灵;fc/b:华蟾酥毒基对蟾毒灵;fc/r:华蟾酥毒基对酯蟾毒配基。
3.2不同柱温和色谱柱流速考察
考察了20、25℃两个柱温和0.5、0.6、0.7mlmin-1三个流速,结果见表7、8。
表7.不同柱温的校正因子(n=6)
fc/a:华蟾酥毒基对沙蟾毒精;fc/t:华蟾酥毒基对远华蟾毒精;fc/bt:华蟾酥毒基对蟾毒它灵;fc/b:华蟾酥毒基对蟾毒灵;fc/r:华蟾酥毒基对酯蟾毒配基
表8.不同流速的校正因子(n=6)
fc/a:华蟾酥毒基对沙蟾毒精;fc/t:华蟾酥毒基对远华蟾毒精;fc/bt:华蟾酥毒基对蟾毒它灵;fc/b:华蟾酥毒基对蟾毒灵;fc/r:华蟾酥毒基对酯蟾毒配基
3.3待测组分色谱峰的定位
利用相对保留时间差定性,知道内标峰的保留时间,加上相对保留时间差,再根据峰形判断,即能够正确判断出目标峰的准确峰位置,见表9。
表9各组分保留时间及相对保留时间差
4.结论
通过方法学确证、方法的耐用性、系统使用考察和检测结果准确性的评价,对本发明蟾酥中化合物的检测方法的技术适用性和应用可行性进行了探讨。结果表明,本发明蟾酥中化合物的检测方法推算的含量与实测法所得含量没有显著性差异,说明试验建立的校正因子可信。可以用于蟾酥质量的定量评价,有望推广其他中药材的多指标质量评价。
上述具体实施方式不能作为对本发明保护范围的限制,对于本技术领域的技术人员来说,对本发明实施方式所做出的任何替代改进或变换均落在本发明的保护范围内。
本发明未详述之处,均为本技术领域技术人员的公知技术。
- 还没有人留言评论。精彩留言会获得点赞!