基于亚细胞定位特异性的关键蛋白质识别方法与流程
本发明属于系统生物学领域,涉及一种基于亚细胞定位特异性的关键蛋白质识别方法。
背景技术:
众所周知,不同的蛋白质对生物有机体生命活动的作用各有不同,重要性也各不相同。一般来说,在蛋白质相互作用网络中维持生物机体生命活动所必需的蛋白质被认为是关键蛋白质。关键蛋白质的缺失将导致细胞死亡或者无法复制繁殖。关键蛋白质的识别可以促进药物标靶技术的发展【注释:药物靶标是指体内具有药效功能并能被药物作用的生物大分子,如某些蛋白质和核酸等生物大分子。那些编码靶标蛋白的基因也被称为靶标基因。事先确定靶向特定疾病有关的靶标分子是现代新药开发的基础。更通俗地讲,就是使用某种药物作用在生物大分子上,影响该生物大分子,从而对疾病产生疗效。这个生物大分子就是药物靶标。】。由于关键蛋白质的剔除、打断或者阻塞都可能导致生物体的死亡。因此,在目前抗菌素普遍存在抗药性和药物毒性问题的背景下,研究一些细菌病毒的关键蛋白质,有助于研发新的抗菌药物。关键蛋白质的识别还有助于合成生物研究的发展。合成生物的目标是找出最小的能执行特定任务的基因组。在2010年,J.CraigVenter学会的研究人员移除丝状支原体的所有非关键基因,创造第一个具有自我复制功能的人造细菌,称作JCVI-syn1.0。目前,识别关键蛋白质的生物实验方法,例如单个基因敲除,RNA干扰和有条件基因敲除等,既浪费时间又效率低下,而且只能在少量物种上实行。因此,迫切需要有效的计算方法来降低实验的成本,提高实验效率。随着高通量的蛋白质相互作用数据的积累,利用蛋白质相互作用网络来识别关键蛋白质成为了目前的研究热点。根据对一些物种的观察,如酵母,线虫和果蝇等,蛋白质相互作用网络中拥有高度的蛋白质结点更有可能是关键蛋白质。从拓扑的角度来看,高度连接的蛋白质结点可以维持蛋白质相互作用网络的基本特征,如果删除这些蛋白质结点将会引起整个蛋白质相互作用网络的崩溃;从生物学的角度来看,这些高度连接的蛋白质结点一般保证了蛋白质相互作用网络的功能完整性。这种现象被认为是生物网络中普遍存在的“中心性-致死性”法则。该法则表明蛋白质相互作用网络中某一蛋白质结点的拓扑特征与蛋白质的关键性之间密切相关。近年来,一些基于网络拓扑特征的中心性方法被提出来识别关键蛋白质,例如度中心性(DegreeCentrality,DC),介数中心性(BetweennessCentrality,BC),接近性中心性(ClosenessCentrality,CC),子图中心性(SubgraphCentrality,SC),特征向量中心(EigenvectorCentrality,EC),信息中心性(InformationCentrality,IC)和邻居中心性(NeighborCentrality,NC)等等。这些方法对蛋白质在相互作用网络中的中心性进行打分,然后将这些得分进行排序来判断蛋白质是否是关键蛋白质。这类方法的优点是不需要预先知道一部分关键蛋白质来训练分类器,而是直接通过给蛋白质打分来预测关键蛋白质。这些基于网络拓扑特征的中心性方法的关键蛋白质预测准确性依赖于蛋白质相互作用网络的可靠性。然而,目前可以得到的蛋白质相互作用网络数据是不完整的,包含很多假阳性和假阴性。这些会影响关键蛋白质识别的准确性。随着高通量生物数据的增多,最近一些研究者试图结合其它生物信息来提高识别关键蛋白质的准确率。例如,Li等人通过考虑基因的功能注释,构建了一个加权的蛋白质相互作用网络,通过集成网络拓扑特征与基因表达信息提出了PeC方法。除此之外,基于机器学习的方法也被用来预测关键蛋白质,常用的特征有GC内容、蛋白质长度、ORF长度等。例如,Acencio等人提出的基于机器学习的方法结合网络的拓扑特征、生物过程信息以及细胞定位等来识别关键蛋白质。这些机器学习的方法通过学习一个物种中一些已知的关键蛋白质的特征,训练出一个分类器,然后利用这个分类器来识别该物种或其它物种中的关键蛋白质。机器学习方法需要预先知道一部分关键蛋白质,其性能取决于分类器的性能以及训练物种与预测物种之间的距离。上述各类方法从不同的角度解决了关键蛋白质识别存在的一些问题。然而,由于可利用的蛋白质相互作用数据存在噪声等因素,基于网络水平的关键蛋白质识别还存在很多挑战。公开号为102176223A的发明专利公开了一种基于关键蛋白质和局部适应的蛋白质复合物识别方法,其核心构思是基于蛋白质相互作用无向图,并以关键蛋白质为种子对关键蛋白质进行识别,这种方法虽然效果较佳,但是受制于种子的选择,因此对于未知的蛋白质识别难度较大。公开号为102841985A的发明专利公开了一种基于结构域特征的关键蛋白质识别方法【申请号为201210282873.7】,其核心构思在于,基于首先通过数据库或进行蛋白质序列分析等得到结构域信息,在结构域信息基础上统计各个结构域类型在蛋白质中出现次数,通过统计结果计算每个蛋白质的权重,权重越大蛋白质成为关键蛋白质的可能性越大。该方法简单有效,且仅基于蛋白质结构域信息,不依赖蛋白质相互作用网络信息等,避免了生物实验所消耗的大量人力物力。通过与随机方法比较,该方法能够较准确的识别关键蛋白质。然而由于结构域信息存在不完整性,会使得预测结果出现偏差。例如,一些的结构域频率低,并不是因为真正具有关键性,而是由于没有测得这些结构域在这个物种中所有蛋白质中的完整分布所致。公开号为101051335公开了一种利用计算机模拟蛋白质相互作用的方法【申请号:200710015493.6】,该方法只是通过计算机软件来模拟蛋白质相互作用,并不涉及到蛋白质的识别。因此,有必要设计一种新型的关键蛋白质识别方法。
技术实现要素:
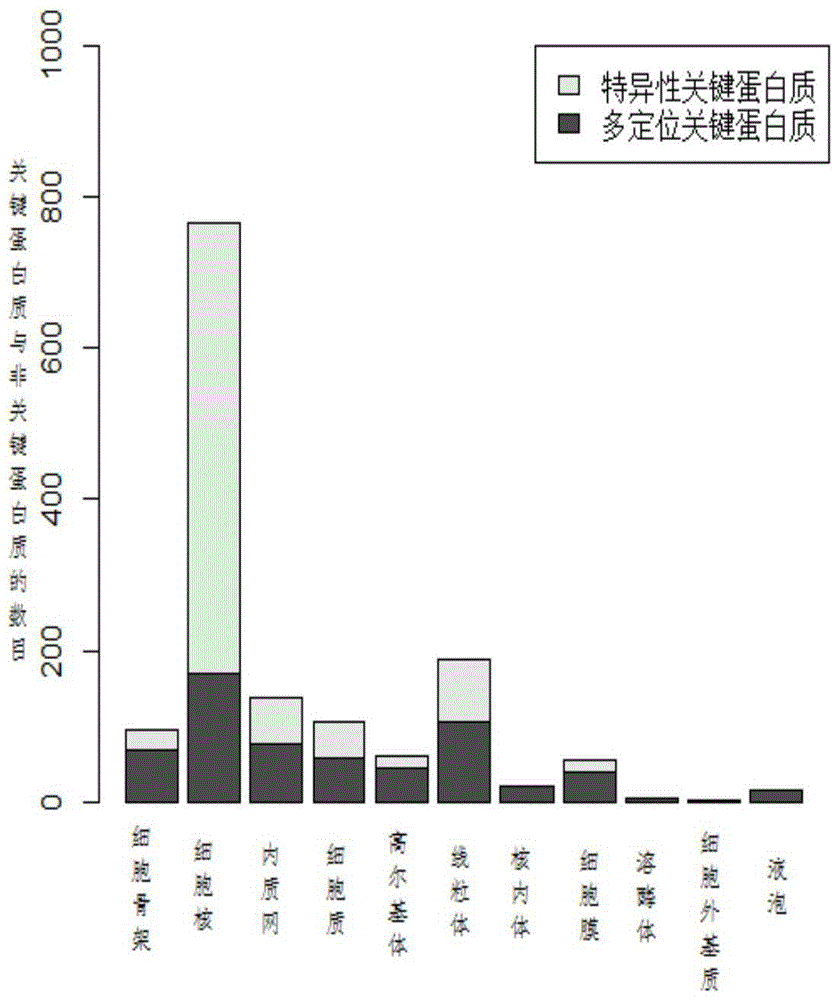
本发明所要解决的技术问题是提供一种基于亚细胞定位特异性的关键蛋白质识别方法,该基于亚细胞定位特异性的关键蛋白质识别方法在关键蛋白质的识别方面准确性高、敏感度高。发明的技术解决方案如下:一种基于蛋白质亚细胞定位特异性的关键蛋白质识别方法,包括以下步骤:步骤1:建立亚细胞定位的蛋白质相互作用子网;输入一组蛋白质的亚细胞定位信息和一组蛋白质相互作用数据,其中蛋白质的亚细胞定位信息包括细胞骨架、细胞核、内质网、细胞质、高尔基体、线粒体、核内体、细胞膜、溶酶体、细胞外基质和液泡共11种亚细胞定位信息;首先在蛋白质相互作用数据中去掉重复相互作用和自相互作用的数据;然后依据亚细胞定位信息以及经过上述处理后的蛋白质相互作用数据构建11个蛋白质亚细胞定位相互作用子网;步骤2:衡量各个蛋白质亚细胞定位相互作用子网的蛋白质关键性得分的可信度;根据各个蛋白质亚细胞定位相互作用子网的规模(子网的规模指子网中包含的蛋白质数目),给每个蛋白质亚细胞定位相互作用子网的蛋白质关键性得分的可信度进行打分,可信度的计算公式如下:其中,|Si|为子网Si中蛋白质的数目,|Smax|为子网Smax中蛋白质的数目,Smax是包含有最多蛋白质的子网;步骤3:计算所有蛋白质的关键性综合得分:将细胞内所有蛋白质的关键性综合得分初始化为0;基于蛋白质在蛋白质亚细胞定位相互作用子网的关键性得分以及不同蛋白质亚细胞定位相互作用子网的蛋白质关键性得分的可信度,按可信度的从高到底依次更新各个子网中的蛋白质的关键性综合得分;关键性综合得分的更新公式为:对于该公式的解释和说明:由于蛋白质p的关键性综合得分C_Ess(p)是基于较高可信度的子网计算的,当前子网的蛋白质关键性得分的可信度小于之前已计算过的子网的蛋白质关键性得分的可信度,因此,更新关键性综合得分的规则为,当蛋白质p的关键性综合得分C_Ess(p)比当前子网Si中p的关键性得分S_Ess(Si,p)高时,其关键性综合得分C_Ess(p)保持不变;而当p的关键性综合得分C_Ess(p)小于当前子网Si中p的关键性得分S_Ess(Si,p)时,关键性综合得分C_Ess(p)更新为原关键性综合得分C_Ess(p)加上当前子网的蛋白质关键性得分S_Ess(Si,p)与原关键性综合得分C_Ess(p)的差值按可信度P(Si)的折算值;其中,S_Ess(Si,p)是采用中心性方法在蛋白质亚细胞定位相互作用子网Si上计算的蛋白质p的关键性得分;P(Si)为子网Si的蛋白质关键性得分的可信度;(例如当采用DC中心性方法时,S_Ess(Si,p)等于蛋白质p在子网Si中连接的边的条数)步骤4:输出结果:对细胞内所有蛋白质按关键性综合得分排序并输出排序结果。不同物种的蛋白质亚细胞定位数据从COMPARTMENTS数据库中获得,蛋白质相互作用数据从公共数据库中获得,所述的公共数据库包括DIP和Biogrid数据库。COMPARTMENTS数据库整合了来自UniProtKB,MGI,SGD,FlyBase,WormBase等数据库的基于实验的蛋白质亚细胞注释信息,涵盖了人类、酵母、果蝇、小鼠等真核生物的蛋白质亚细胞定位信息。DIP和Biogrid等公共数据库里包含了许多物种的蛋白质相互作用数据。有益效果:本发明基于对关键蛋白质在不同亚细胞定位的分布的差异性和特异性,提出了一种基于蛋白质亚细胞定位特异性的关键蛋白质识别方法(LSED),利用蛋白质亚细胞定位数据和相互作用数据构建了蛋白质亚细胞定位相互作用子网,分别对每个子网中的蛋白质进行关键性打分,通过对蛋白质亚细胞定位相互作用子网的蛋白质关键性得分的可信度进行衡量,并结合各个蛋白质亚细胞定位相互作用子网进行蛋白质的关键性预测来计算蛋白质的关键性综合得分。本发明简单易用,试验表明,与已有的基于网络拓扑特征的关键蛋白质识别方法相比较,本发明提出的方法在预测的准确性、与已知关键蛋白质匹配的敏感度和特异性等方面都有明显提高,能为生物学家进行关键蛋白质识别的实验和进一步研究提供有价值的参考信息。附图说明图1:各个蛋白质亚细胞定位相互作用子网中关键蛋白质和非关键蛋白质的数目图2:各个蛋白质亚细胞定位相互作用子网中多定位关键蛋白质以及特异性关键蛋白质的数目比图3:本发明LSED的流程图;图4:在酵母蛋白质相互作用网络上不同比例的具有较高排序得分的蛋白质作为预测的关键蛋白质时,方法LSED和6种拓扑中心性方法识别关键蛋白质的数量的比较图;图a-f分别是比例为1%、5%、10%、15%、20%、25%时的对比图。图5:方法LSED和6种拓扑中心性方法与LSED-NC的ROC曲线的比较图;图a-f分别为DC与LSED-DC,IC与LSED-IC,EC与LSED-EC,SC与LSED-SC,BC与LSED-BC,NC与LSED-NC的ROC曲线的比较图;图6:方法LSED和6种拓扑中心性方法基于jackknife曲线的比较图;图6(a)-(f)分别展示的是DC与LSED-DC,IC与LSED-IC,EC与LSED-EC,SC与LSED-SC,BC与LSED-BC,NC与LSED-NC的jackknife曲线比较结果。图7:方法LSED和6种拓扑中心性方法识别的蛋白质重叠性和差异性比较;具体实施方式以下将结合附图和具体实施例对本发明做进一步详细说明:实施例1:(1)蛋白质亚细胞定位相互作用子网构建及关键蛋白质的分布特性分析不同物种的蛋白质亚细胞定位数据可以从COMPARTMENTS数据库中获得。COMPARTMENTS数据库整合了来自UniProtKB,MGI,SGD,FlyBase,WormBase等数据库的基于实验的蛋白质亚细胞注释信息,涵盖了人类、酵母、果蝇、小鼠等真核生物的蛋白质亚细胞定位信息。DIP和Biogrid等公共数据库里包含了许多物种的蛋白质相互作用数据。目前研究最为广泛的物种是酵母,其蛋白质相互作用网络和关键蛋白质数据在众多物种中是最为完整和可靠的,因此,首先基于酵母的数据进行数据分析和实验验证。酵母的蛋白质相互作用网络来自于DIP数据库2010年10月的数据。除去自相互作用和重复的相互作用,总共有5093个蛋白质,24743条边。酵母蛋白质亚细胞定位注释信息来自COMPARTMENTS数据库,酵母蛋白质分别被11种亚细胞定位注释,其中包括细胞骨架、细胞核、内质网、细胞质、高尔基体、线粒体、核内体、细胞膜、溶酶体、细胞外基质和液泡等亚细胞定位。关键蛋白质信息来源于MIPS,SGD,DEG和SGDP四个数据库。在5093个蛋白质中1167个为关键蛋白质,剩余3926个蛋白质视作非关键蛋白质。将DIP数据库里的酵母蛋白质相互作用网络分别映射到每个亚细胞定位,一共构建了酵母的11个蛋白质亚细胞定位相互作用子网。每个子网由被这种亚细胞定位注释的蛋白质以及它们之间的相互作用构成。经过这种映射,包含分别位于两个不同的亚细胞定位的蛋白质的相互作用被丢掉了,这使得任何一个蛋白质亚细胞定位相互作用子网中相互作用的蛋白质都处于同一亚细胞定位,从而保证了蛋白质相互作用的发生。我们统计了各个蛋白质亚细胞定位相互作用子网的蛋白质分布以及关键蛋白质分布,如图1所示。从图1可以看出,不同蛋白质亚细胞定位相互作用子网的蛋白质数量以及关键蛋白质数量存在很大的差异性。其中,处于蛋白质细胞核(Nulceus)亚细胞定位相互作用子网的蛋白质数量以及关键蛋白质数量都显著高于其他蛋白质亚细胞定位相互作用子网。从图1还可以发现,不同蛋白质亚细胞定位相互作用子网的关键蛋白质与非关键蛋白质的比例存在较大差异;关键蛋白质在蛋白质细胞骨架(Cytoskeleton)、细胞核(Nulceus)、内质网(Endoplasmic)、细胞质(Cytosol)、以及高尔基体(Golgi)亚细胞定位相互作用子网中的比例高于关键蛋白质在DIP数据库里的酵母蛋白质相互作用网络中的比例。分析的结果表明:关键蛋白质在各个蛋白质亚细胞定位相互作用子网的分布具有显著的统计特性,且关键蛋白质主要在某些亚细胞定位相互作用子网富集,这说明并非所有的蛋白质亚细胞定位相互作用子网的蛋白质关键性评分都对细胞内关键蛋白质的预测有用。(2)统计分析关键蛋白质的亚细胞定位特异性为考察关键蛋白质的亚细胞定位特异性,统计各个蛋白质亚细胞定位相互作用子网中特异性关键蛋白质(只出现在某个蛋白质亚细胞定位相互作用子网中的关键蛋白质)和多定位关键蛋白质(出现在至少两个蛋白质亚细胞定位相互作用子网中的关键蛋白质)占各个蛋白质亚细胞定位相互作用子网中关键蛋白质的比例,统计结果如图2所示。从图2中,我们可以发现在蛋白质细胞核(Nulceus)亚细胞定位相互作用子网中,具有亚细胞特异性的关键蛋白质占主要成分。另外,在蛋白质核内体(Endosome)定位亚细胞相互作用子网和蛋白质液泡(Vacuole)亚细胞定位相互作用子网中,特异性关键蛋白质非常少。这说明这些蛋白质亚细胞定位相互作用子网中几乎所有的关键蛋白质能够通过其他蛋白质亚细胞定位相互作用子网识别。在1167个关键蛋白质中,包括了852个特异性关键蛋白质和315个交叠性关键蛋白质,由此可知,大多数关键蛋白质具有亚细胞定位特异性。因此分别在各个蛋白质亚细胞定位相互作用子网中预测关键蛋白质,能较容易地够检测到特异性关键蛋白质,从而能够提高关键蛋白质预测的准确性。(3)基于亚细胞定位特异性的蛋白质关键性综合得分计算基于蛋白质亚细胞定位特异性的关键蛋白质识别方法(LSED)的流程图如图3所示。首先,将蛋白质相互作用网络分别映射到每个亚细胞定位,一共构建了若干个蛋白质亚细胞定位相互作用子网。在各个蛋白质亚细胞定位相互作用子网中采用中心性方法,对子网中各个蛋白质的关键性进行打分。然而,由于各个子网的网络拓扑不同,多定位蛋白质(出现在至少两个蛋白质亚细胞定位相互作用子网中的蛋白质)在不同的蛋白质亚细胞定位相互作用子网中关键性评分也必然存在差异;另一方面,由于不同的蛋白质亚细胞定位相互作用子网规模等存在差异,特异性蛋白质(只出现在某个蛋白质亚细胞定位相互作用子网中的蛋白质)为关键蛋白质的可能性高低也不能简单通过唯一的关键性评分来决定。我们基于多个蛋白质亚细胞定位相互作用子网,计算每个蛋白质的关键性综合得分,具体步骤如下:步骤1考虑到不同的蛋白质亚细胞定位相互作用子网中预测关键蛋白质的准确度存在差异性,我们对所有的蛋白质亚细胞定位相互作用子网的蛋白质关键性得分的可信度进行衡量。从上述分析可以发现,蛋白质亚细胞定位相互作用子网规模越大(网络规模指网络中包含蛋白质的数目),特异性蛋白质数目越多,包含的关键蛋白质数目也越多,从而蛋白质关键性得分的可信度越高。假设Smax是网络规模最大的蛋白质亚细胞定位相互作用子网,相比于其他子网,该子网蛋白质关键性得分的可信度最高。对于其他蛋白质亚细胞定位相互作用子网Si,通过考察该子网的规模与Smax的网络规模的比值,可信度计算如公式1所示。其中,|Si|为子网Si中蛋白质的数目,|Smax|为子网Smax中蛋白质的数目,Smax是包含有最多蛋白质的子网。步骤2采用一种中心性方法(DC,IC,EC,SC,BC或NC),分别在每个蛋白质亚细胞定位相互作用子网上计算各个蛋白质的关键性得分。假设蛋白质p存在于蛋白质亚细胞定位相互作用子网Si中,计算的关键性得分标记为S_Ess(Si,p)。步骤3计算所有蛋白质的关键性综合得分:一个蛋白质可能出现在不同的蛋白质亚细胞定位相互作用子网中,从而有多个关键性得分,因此需要为每个蛋白质计算关键性综合得分来衡量它的关键性。计算所有蛋白质的关键性综合得分步骤:1)将细胞内所有蛋白质的关键性综合得分初始化为0;2)根据子网的蛋白质关键性得分的可信度从高到底,依次更新各个子网中蛋白质的关键性综合得分。由于蛋白质p的关键性综合得分C_Ess(p)是基于较高可信度的子网计算的,当前子网的蛋白质关键性得分的可信度小于之前已计算过的子网的蛋白质关键性得分的可信度,因此,更新关键性综合得分的规则为,当蛋白质p的关键性综合得分C_Ess(p)比当前子网Si中p的关键性得分S_Ess(Si,p)高时,其关键性综合得分C_Ess(p)保持不变;而当p的关键性综合得分C_Ess(p)小于当前子网Si中p的关键性得分S_Ess(Si,p)时,关键性综合得分C_Ess(p)更新为原关键性综合得分C_Ess(p)加上当前子网的蛋白质关键性得分S_Ess(Si,p)与原关键性综合得分C_Ess(p)的差值按可信度P(Si)的折算值。关键性综合得分的更新规则如公式2所示。例如,当轮到可信度第i高的蛋白质亚细胞定位相互作用子网Si时,对子网中的每个蛋白质p,比较其在子网Si中的关键性得分S_Ess(Si,p)和其关键性综合得分C_Ess(p),将其关键性综合得分C_Ess(p)按公式2进行更新。其中,otherwise为否则的意思。当所有的蛋白质亚细胞定位相互作用子网中的蛋白质都依次检查完毕,对细胞内所有蛋白质按关键性综合得分进行降序排序。最后关键性综合得分越高的越可能为关键蛋白质。(4)基于蛋白质亚细胞定位特异性的关键蛋白质识别方法(LSED)的有效性验证为了评估LSED方法的有效性,我们将LSED方法分别与几种代表性的中心性方法结合,如DC,BC,SC,EC,IC,以及NC,计算酵母所有蛋白质的关键性综合得分,按降序排序;另外,只用按中心性方法对酵母的蛋白质相互作用网络中的蛋白质进行关键性打分,按降序排序,作为对照试验。酵母的蛋白质相互作用网络来自于DIP数据库2010年10月的数据,包含有5093个蛋白质,24743条边。酵母蛋白质亚细胞定位注释信息来自COMPARTMENTS数据库,将DIP数据库里的酵母蛋白质相互作用网络分别映射到每个亚细胞定位,一共构建了酵母的11个蛋白质亚细胞定位相互作用子网。来源于MIPS,SGD,DEG和SGDP四个数据库的1167个关键蛋白质作为标准集,用来比较预测结果的准确性。a.与六种中心性方法比较选取排在前面1%,5%,10%,15%,20%,25%的蛋白质作为预测的关键蛋白质。再依据已知的关键蛋白质集合,对比预测结果。预测正确的蛋白质数量作为评价各个方法性能的标准。这种评价方法已经被以前的研究广泛采用。我们选择不同比例的具有较高排序得分的蛋白质作为预测的关键蛋白质,然后比较LSED方法和中心方法识别关键蛋白质的数量。图4表明,相比于各种中心性方法(总称XC)的预测结果,结合不同中心性方法的LSED方法(总称LSED-XC)极大提高了正确预测的关键蛋白质数量。当选择排序得分排在前1%的蛋白质作为预测的关键蛋白质时,LSED方法的准确率范围64%-80%左右,而各种中心性方法的准确率最高仅为62%。当选择排序得分排在前25%的蛋白质作为预测的关键,LSED-XC可以达到47.1%的准确率,而中心性方法中准确率最高为42.7%。NC是在现存的中心性方法中性能最好的中心性方法。与NC相比,在选择排序得分排在前1%,5%,10%,15%,20%和25%的蛋白质作为关键蛋白质时,LSED-NC预测的准确率能够分别提高25%,15%,9.2%,12.3%,10.9%,10.9%。DC是一种广泛使用的关键蛋白质的预测方法。与DC相比,LSED-DC也体现了明显的优势。特别是在选前1%的蛋白质时,LSED-DC预测关键蛋白质的准确性是DC的1.77倍。在选择前5%和前10%的蛋白质时,LSED-DC预测关键蛋白质的准确性比DC提高了56.4%和42.9%。相比于其他中心性方法(XC),LSED-XC的优势也非常明显。我们可以发现,与任意一种中心性方法结合,LSED方法预测关键蛋白质的准确性高于绝大多数的中心性方法的准确性。b.基于ROC曲线比较实验结果用ROC曲线以及其下的面积(AUC)进一步验证LSED方法和各种中心性方法的性能。将细胞中所有的蛋白质依据各个方法计算的关键性得分降序排序,排在前面的K个蛋白质作为预测的关键蛋白质(阳性数据集),细胞中剩下的蛋白质作为非关键蛋白质(阴性数据集)。阈值K的范围从1到5093。随着K取值的不同,分别计算各个方法相应的Sensitivity和Specificity值,从而得到ROC曲线,如图5所示。图5(a)-(f)分别显示了是DC与LSED-DC,IC与LSED-IC,EC与LSED-EC,SC与LSED-SC,BC与LSED-BC,NC与LSED-NC的ROC曲线对比图。从图5,我们可以看出各种LSED-XC的ROC曲线明显高于各种XC中心性方法的ROC曲线。另外,我们还可以观察到,各种LSED-XC的AUC明显大于各种XC中心性方法的AUC。c.基于jackknife曲线比较实验结果我们用jackknife曲线来进一步验证LSED方法以及其他XC方法的预测性能。实验结果如图6所示。X轴表示的是按照各个关键蛋白质预测方法排序,排在前面的蛋白质数量。Y轴表示的是识别的正确关键蛋白质的累积数量。图中线下的面积用来比较各个方法的性能。图6(a)-(f)分别展示的是DC与LSED-DC,IC与LSED-IC,EC与LSED-EC,SC与LSED-SC,BC与LSED-BC,NC与LSED-NC的jackknife曲线比较结果。从图6,我们可以看出各种LSED-XC对应的jackknife曲线位于相应的XC对应的jackknife曲线的上方,曲线下的面积明显大于相应的XC对应的曲线下的面积。所有的实验结果表明LSED方法预测关键蛋白质的准确性高于各种中心性的方法。d.LSED与其他中心性方法在预测上的差异比较被各个方法排在前100位的蛋白质,计算它们的重叠预测和差异预测。如图7所示,LSED-XC识别的排在前100位的蛋白质中,有一半以上与相应的XC方法识别的蛋白质不同。这说明LSED方法的亚细胞特异性信息是导致这种不同的主要原因。为了进一步的分析,我们比较了LSED方法和中心性方法识别的不同蛋白质中关键蛋白质的比率。如图7所示,对比各种XC和LSED-XC方法识别的不同蛋白质,LSED-XC能够找到更多的不同的关键蛋白质。以NC为例,在前100个蛋白质中LSED-NC找到了56个不同的蛋白质,其中48(48/56=85.7%)个是关键蛋白质,而仅有19(48/56=33.9%)个不同关键蛋白质被NC找到而被LSED-NC忽略。这说明,通过基于蛋白质亚细胞定位相互作用子网的关键性综合评分比基于全局蛋白质相互作用网络的关键性评分更加准确,基于亚细胞定位特异性的蛋白质关键性综合得分更能刻画蛋白质的关键性,且能够降低了假阳性相互作用对中心性计算造成的影响。综上所述,基于本发明的基于蛋白质亚细胞定位特异性的关键蛋白质识别方法(LSED),亚细胞定位信息对预测出来的关键蛋白质的准确性以及与已知关键蛋白质匹配的敏感度和特异性等方面具有重要作用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1