一种析氢电极材料合金成分的筛选方法与流程
本发明涉及一种电极材料合金成分的筛选方法,特别涉及一种析氢电极材料合金成分的筛选方法,属于电极材料
技术领域:
。
背景技术:
:计算材料学是一门正在快速发展的新兴学科,它可以通过计算机对材料组成、结构、性能等多方面进行模拟和设计。模拟的工作仅在计算机上即可完成,因此基本不受实验条件、时间和空间的限制,具有极强的灵活性和随机性。通常的新材料设计需要大量的实验工作进行反复验证,而现在这些实验过程越来越多的可以直接通过计算机模拟完成,从而节省大量的人力和物力,并加快新材料的研发。计算材料学在材料设计与开发领域的应用必将推动材料科学及相关产业的快速发展。氢气作为二次能源具有清洁无污染、燃烧热值高、可存储和运输等优点,受到世界各国广泛重视。目前,通过电解水制氢是实现工业化廉价制备氢气的重要方法。传统的阴极材料为铁或镀镍阴极,但由于具有较高的析氢过电位,能耗大,严重地阻碍电解水工业的发展。为了降低能耗,研究开发新型的、廉价的、高催化活性的析氢电催化材料,更加具有商业价值和现实意义。影响阴极材料活性的因素主要可以分为几何因素和能量因素。几何因素主要考虑材料的比表面积和表面形貌特征,很大程度取决于制备工艺;能量因素即以金属-氢键的键能为判断依据。目前比较流行的析氢理论是迟缓放电理论和复合理论。通常认为在碱性或中性介质中的电极反应基本过程如下:[1]电化学反应:产生吸附在电极表面的氢原子h20+e+m—mh+oh-;[2]复合脱附:mh+mh—2m+h2;[3]电化学脱附:mh+h20+e—h2+m+oh-。以上析氢过程表明高活性的析氢电极,不仅要求在析氢电催化反应过程中容易形成金属-氢键,而且还要求被吸附物具有良好的解吸附能力。对于基体材料m,m-h键的键强度较强时容易发生氢吸附;m-h键的键强度较弱时容易发生氢脱附。 因此,筛选出具有适量吸附氢特性的金属与基体m形成活性较高的合金,可以显著地提高析氢电化学反应速率。实验对析氢过程研究已很成熟,大量的实验表明金属-氢键键能与析氢反应电流之间呈现“火山型”关系图。在计算材料领域中,量子力学第一原理方法已广泛应用于析氢电催化反应,并且已经与实验建立了联系。理论计算表明氢的吸附自由能δgh可以作为催化反应的描述符,它是一个关联实验的金属-氢键键能物理量,并且与析氢反应电流之间保持着“火山型”关系图。当δgh越接近于0时,析氢反应的交换电流密度越大,也就是说δgh=0的材料是催化活性最高的催化剂。因此,计算模拟可以通过氢的吸附自由能筛选合适的电极材料。大量的实验表明电极活性与m-h键能密切相关,但是由于实验局限性,研究往往限于纯金属体系。将计算材料学用于析氢电极材料合金成分的筛选将有助于该
技术领域:
的进一步发展。因此,提供一种简单快捷地筛选出能够提升基体材料电极催化活性的合金元素的方法就成为该
技术领域:
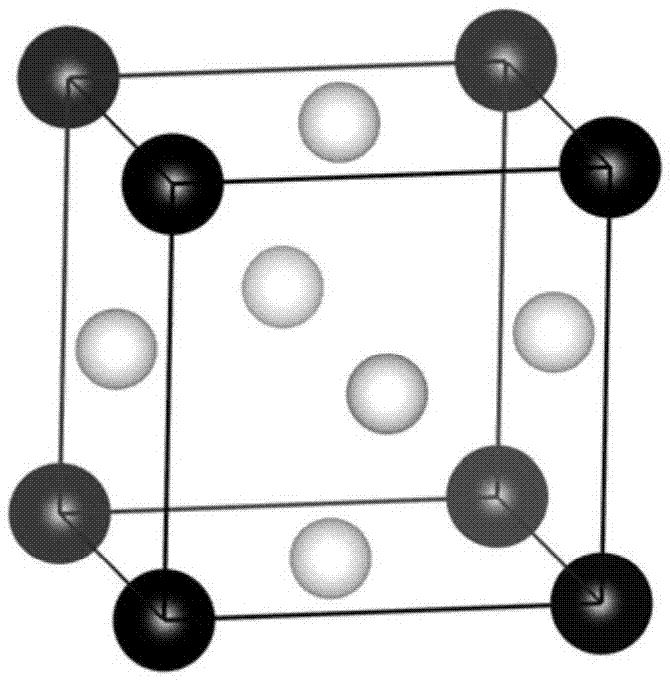
急需解决的难题。技术实现要素:本发明的目的在于提供一种析氢电极材料合金成分的筛选方法,采用该方法可以简单快捷地筛选出能够提升基体材料电极催化活性的合金元素,与传统实验方法相比,大幅度削减了实验费用,降低了新材料的研发周期。为实现上述目的,本发明采用以下技术方案:一种析氢电极材料合金成分的筛选方法,包括以下步骤:(1)计算基体及合金的晶格常数:通过量子力学第一原理方法计算基体的晶格常数,与此同时,在基体原胞中通过替代方式放入掺杂合金元素,从而构建出合金原胞,并用相同方法获得合金晶格常数;(2)模拟基体及合金表面氢吸附过程:利用基体及合金原胞参数构建基体表面模型和合金表面模型,并通过量子力学第一原理方法模拟氢吸附过程,计算氢原子在基体表面和合金表面的吸附能δeh;(3)关联计算模拟与实验:以往的实验结果表明金属-氢键能与析氢反应电流之间呈“火山型”关系;同时,理论计算证实表面氢的吸附自由能δgh是一个与实验上金属-氢键能关联的物理量,将其作为催化反应的描述符,根据δgh=δeh+0.24,获得基体和合金表面氢的吸附自由能;(4)建立析氢电极催化活性数据库:针对不同的合金元素,重复以上步骤,最终筛选出高催化活性的合金电极并建立析氢电极催化活性数据库。优选地,所述步骤(2)中构建合金表面模型时考虑掺入元素引起的晶格变化,基体原胞中部分原子被掺杂元素替代,数目不能超过50%,晶格常数精度至少为优选地,所述步骤(2)中表面模型的层厚至少为4层原子,真空层厚度至少为优选地,所述步骤(1)中所述基体为镍,所述掺杂合金元素为mo、fe、co、cr、sn、la、ti、sc、y、cu或hf。在本发明的筛选方法中,合金电极的筛选主要以能量因素即金属-氢键的键能为判断依据,通过键能筛选出具有适量吸附氢特性的金属与基体形成活性较高的合金,从而达到提升电极材料的催化性能的效果。本发明的优点:本发明一种新型的析氢电极材料合金成分的筛选方法,利用该方法能够合理有效地筛选出掺入的合金元素,用于提升电极材料的催化性能。该方法借助于量子力学第一原理计算直接在计算机上模拟合金的析氢过程,很大程度减少了传统实验方法人力物力的支出,明显地提高了研发效率。该析氢电极材料合金成分的筛选方法具有很强的实用价值,它不仅仅应用于析氢反应,也适用与其它反应中催化剂的合金化设计。下面通过附图和实施例对本发明作详细说明,但并不意味着对本发明保护范围的限制。附图说明图1-1为本发明实施例1中ni基体原胞结构示意图,图1-2为本发明实施例1中ni3mo合金原胞结构示意图。图2-1为本发明实施例1中氢吸附ni3mo合金(111)表面侧视图;图2-2为本发明实施例1中氢吸附ni3mo合金(111)表面俯视图。图3为m-h键能与析氢反应电流之间的“火山型”关系图。具体实施方式为本发明实施步骤如下:首先,计算基体及合金的晶格常数,接着构建基体及合金表面模型,模拟氢吸附过程并计算氢原子在基体及合金表面的吸附能,然后关联计算模拟与实验,获得衡量催化活性的物理量(氢吸附自由能δgh),最后筛选出具有高催化活性的合金电极并建立合金电极催化活性数据库。实施例1以ni为例,ni基电极材料合金成分的筛选方法,包括如下步骤:1、计算ni及其合金的晶格常数:首先建立ni的计算模拟原胞,如图1-1所示,为本发明实施例中ni基体原胞结构示意图。利用量子力学第一原理模拟计算,可以获得ni的晶格常数为接着,在基体原胞中通过替代式掺杂放入少量的合金元素mo,形成ni3mo合金,结构如图1-2所示,本发明实施例中ni3mo合金原胞结构示意图,通过计算模拟,合金的晶格常数为2、模拟ni基体及合金表面氢吸附过程:对于ni基电极,发生析氢反应的表面为(111)面,因此,在模拟中建立的表面模型为(2×2)的ni(111)拓展表面,原子共8层,真空层厚度为氢原子被放置于面心立方结构的金属ni表面最稳定的占据位置,氢原子在基体和合金表面的吸附能公式如下:e(surf+nh)为n个氢原子吸附在表面的总能量,e(surf)为没有氢吸附的纯净表面的能量,e(h2)为氢分子的能量,模拟计算结果表明ni基电极表面氢的吸附能δeh为-0.67ev;对于ni-mo合金体系,采用相同的方法建立合金表面,模型如图2-1和图2-2所示,利用该模型进行量子力学第一原理计算,可以获得ni-mo合金电极表面氢的吸附能δeh为-0.47ev;3、关联计算模拟与实验:以往的实验结果表明金属-氢键能与析氢反应电流之间呈“火山型”关系,如图3所示;同时,理论计算的研究工作也证实表面氢的吸附自由能δgh是一个与实验上金属-氢键能关联的物理量,可以作为催化反应的描述符,根据δgh=δeh+0.24,获得在ni基体及合金表面氢的吸附自由能分别为-0.43ev和-0.23ev,如表1所示。4、选择其它种类元素掺入形成合金,重复以上步骤,筛选性能优越的合金电极并建立析氢电极催化活性数据库:以mo为例,选择其它掺入元素,如fe、co、cr、sn、la、ti、sc、y、cu和hf等进行计算模拟,合金的晶格常数及氢的吸附自由能数值结果在表1中列出。实验中以金属-氢键能为衡量电极材料催化活性的指标,m-h键能与析氢反应电流之间呈“火山型”关系,如图3所示,m-h键能与析氢反应电流之间的“火山型”关系图。图3中可以看出:对于某特定的金属-氢键的键能,析氢反应电流最大,电极材料的催化活性最高。在计算模拟中,催化反应的描述符为氢吸附自由能δgh。它与析氢反应电流之间也呈现“火山型”关系,且当δgh=0时,电极材料的催化活性最高。ni的吸附自由能为-0.43ev,因此δgh>-0.43的合金可 以提升电极材料的催化性能。比较表1的数值可以发现mo、fe、co、cr、sn、la和ti为合适的掺入元素,sc、y、cu和h则不合适。实验上已经制备出ni-mo、ni-fe、ni-co、ni-cr、ni-sn、ni-la和ni-ti析氢电极。此计算结果可以存储为析氢电极催化活性数据库,为实验提供数据支持。利用本发明,可以快捷地筛选出具有适量吸附氢特性的金属与基体形成活性较高的合金,有利于新型电极催化材料的研制和设计,很大程度上减少人力和物力的投入。表1:ni基体及多种合金的晶格常数a0(单位)和表面氢的吸附自由能δgh(单位ev)nimofecocrsnlatiscycuhfa03.523.643.503.523.553.743.963.613.723.873.543.73δgh-0.43-0.23-0.31-0.38-0.24-0.35-0.35-0.40-0.47-0.44-0.45-0.44本发明的析氢电极材料合金成分的筛选方法,通过计算基体及合金的晶格常数,构建它们的表面并模拟氢吸附过程,然后获取氢原子在表面的吸附能数据,接着通过关联计算模拟与实验,即可得到计算模拟中衡量催化活性的物理量(氢吸附自由能δgh),进而筛选出具有高催化活性的合金电极。计算模拟的数据可存储为合金电极催化活性数据库,用于实验中对于掺入基体的合金元素进行筛选。该方法结合先进的材料模拟计算能够方便快捷地开发新型廉价的、高催化活性的析氢电催化材料,具有重要的理论和实际价值。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1