一种基因组变异检测方法及检测装置与流程
本发明实施例涉及生物信息研究领域,尤其涉及一种基因组变异检测方法及检测装置。
背景技术:
随着基因组测序成本的持续下降,高通量测序仪(如:Illumina Hiseq系列测序仪)产生的基因组测序序列(read)呈现爆照式增长,尤其是人类基因组测序序列的积累速度更为明显,如何从大量基因组测序序列得到高质量的基因组变异结果,成为一项富有挑战性的工作。
目前,人们通常利用短序列比对软件(如:Burrows Wheeler Aligner,BWA)将每条测序序列比对到参考序列(reference sequence)上,得到每条测序序列与参考序列的双序列比对结果(包括测序序列相对于参考序列详细的匹配、错配、插入和删除等信息),然后根据所有测序序列与参考序列的双序列比对结果,得到基因组变异结果。但由于BWA等短序列比对软件是把每个read单独比对到参考序列上,并不考虑测序序列之间整体上是否对齐,很容易把原本属于同一种变异类型的测序序列错误地比对成包含不同变异类型的测序序列,造成误判。
为解决该问题,现有技术人员通常采用基因分析工具(英文:The Genome Analysis Toolkit,GATK)中的子模块HaplotypeCaller(简称:HC)来进行基因组变异检测,以消除测序序列间比对不齐造成的影响。其中,HC检测主要包括下述4个步骤:1)确定基因组中的潜在变异区域(ActiveRegion);2)对每个潜在变异区域执行局部组装(local assemby),根据组装后的序列以及kmer长度(Size)得到DeBruijn图(DeBruijn graph),并对DeBruijn graph进行处理得到至少一个单倍体(haplotype);3)利用PairHMM计算每个read与每个haplotype的最佳比对、以及每个read与每个haplotype对应的条件概率P(read|haplotype);4)对每个“ActiveRegion”中的每个潜在变异位置,基于二倍体假设,计算出贝叶斯后验概率Pr(D|G)最大的一对haplotypes,将该对haplotypes作为该潜在变异位置的变异结果,其中,G表示基因型(Genotype),D表示覆盖潜在变异区域中的每个变异位置的测序序列。
虽然HC检测能有效避免测序序列间不对齐造成的影响,但仍存在下述几种问题:1)由于在上述local assembly阶段使用了多个不同的kmer size,且对每个不同的kmer size分别建立一张DeBruijn graph并独立地遍历生成haplotypes,所以会导致haplotypes数目偏多,进而导致haplotypes跟参考序列进行Smith-Waterman(简称SW)比对的次数偏多,影响了运行时间。2)局部组装精度不够高;3)由于是基于二倍体假设,所以只支持二倍体变异,不支持多倍体变异,无法很好地支持多倍体变异检测;4)即便在二倍体情况下,计算出的Pr(D|G)也不够精准,例如:在二倍体假设下,覆盖每个变异位置的测序序列,应该是一部分来自于H1,剩下一部分来自于H2,且每个测序序列间相互独立,所以Pr(D|G)的最大值应该是来自于覆盖每个变异位置的测序序列的最佳分割:一部分来自H1,剩下一部分来自H2,且连乘的积最大,然而,GATK只是通过下述计算公式,通过计算均值的连乘来近似得到Pr(D|G):
所以,由上可知现有HC基因组变异检测方法运行时间较长,效率低下,且检测精度不高。
技术实现要素:
为解决上述问题,本发明提供一种基因组变异检测方法及装置,以解决现有基因组变异检测效率低下、且检测精度不高的问题。
为达到上述目的,可选的,本发明的实施例采用如下技术方案:
第一方面,本发明提供一种基因组变异检测方法,由检测装置执行,该方法可以包括:
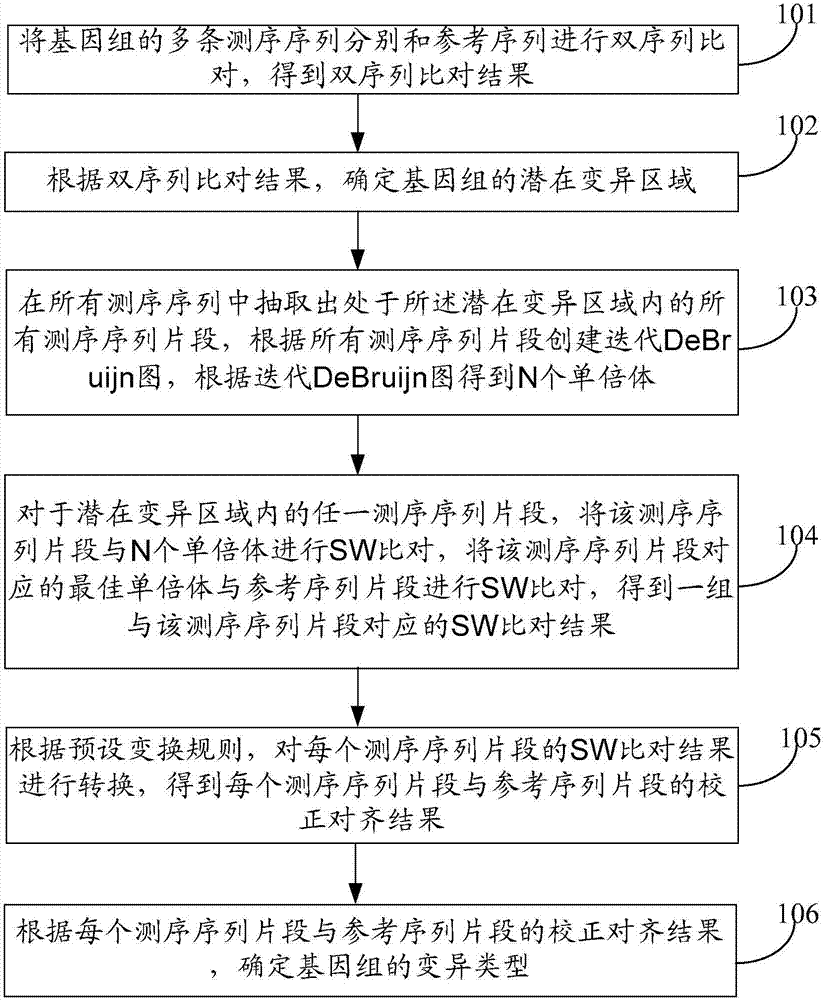
将基因组的多条测序序列分别和参考序列进行双序列比对,得到双序列比对结果,根据双序列比对结果,确定基因组的潜在变异区域,对每个潜在变异区域,在所有测序序列中抽取出处于潜在变异区域内的所有测序序列片段,根据所有测序序列片段创建迭代DeBruijn图,根据迭代DeBruijn图得到N个单倍体;对于潜在变异区域内的任一测序序列片段,将测序序列片段与N个单倍体进行SW比对,将测序序列片段的最佳单倍体与参考序列片段进行SW比对,得到一组与该测序序列片段对应的SW比对结果,根据预设变换规则,对每个测序序列片段的SW比对结果进行转换,得到每个测序序列片段与参考序列片段的校正对齐结果;预设变换规则用于将每个测序序列片段与参考序列片段对齐,根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型。
如此,可以使用局部组装创建创建单张迭代DeBruijn图,减少了DeBruijn图的创建个数,进而减少单倍体的个数,使得减少单倍体与参考序列进行SW比对的次数大大降低,提升了比对速度;其次,本案通过先把每个测序序列片段定位到其比对最佳的单倍体上,然后借助该单倍体与参考序列片段间的比对结果,根据预设转换规则,把该测序序列片段校正对齐到参考序列上,实现了测序序列片段的整体对齐,便于把具有相同变异类型的测序序列片段聚在一起对齐,避免将属于同一类型的变异错误地比对成不同类型的变异,从而提高基因组变异检测结果的准确性。
在第一方面的一种可实现方式中,结合第一方面,可以采用基因分析工具GATK中子模块HaplotypeCaller基于熵的过滤策略来根据双序列比对结果,确定基因组的潜在变异区域,具体如下:
根据基因组的编码顺序,将基因组划分为多个编码区间,根据双序列比对结果,确定所有测序序列的变异类型,统计每个编码区间内不同变异类型的测序序列的概率分布值,根据编码区间内不同变异类型的测序序列的概率分布值,计算编码区间的信息熵,判断每个编码区间的信息熵是否大于第一阈值,若存在第一编码区间,第一编码区间的信息熵大于第一阈值,则确定第一编码区间为潜在变异区域。
还可以采用其他的自定义的启发式策略确定基因组的潜在变异区域,其具体实现可以包括以下步骤:
根据基因组的编码顺序,将基因组划分为多个编码区间,依次统计每个编码区间内发生变异的测序序列的数量,判断每个编码区间内发生变异的测序序列的数量是否大于第二阈值,若存在第一编码区间,其内发生变异的测序序列的数量大于第二阈值,则确定该第一编码区间为潜在变异区域。
如此,可以通过上述任一种方式确定基因组的潜在变异区域。
在第一方面的又一种可实现方式中,结合第一方面或者第一方面的任一种可实现方式,可以采用下述方式创建迭代DeBruijn图:
a)初始化kmer的值k;
b)从所有测序序列当中过滤掉支持数低于设定阈值的kmer,并以kmer作为节点,过滤后的测序序列上的相邻kmer作为边,创建最初始的DeBruijn图;
c)遍历当前DeBruijn图产生多个contig,过滤出长度大于测序仪读长的contig作为新扩充的测序序列,并删除掉所有测序序列中被新扩充的测序序列完全包含的测序序列,同时,将k值加1;
d)以所有测序序列中剩下的测序序列和新扩充的测序序列按照新的k值,以kmer作为节点,所有测序序列上的相邻kmer作为边创建新的DeBruijn图;其中,k值为kmer长度,当前DeBruijn图根据k值创建;
重复上述c)~d)过程,直至k值为预设的最大kmax值,将最大kmax值对应的当前DeBruijn图作为最终需要的迭代DeBruijn图。
如此,可以根据上述方式创建单张迭代DeBruijn图,与现有技术相比,降低了DeBruijn图的创建个数,进而减少了单倍体的个数。
在第一方面的再一种可实现方式中,结合第一方面或者第一方面的任一种可实现方式,对于任一测序序列片段的SW比对结果,SW比对结果可以包括:该测序序列片段与最佳单倍体进行SW比对后的第一SW比对结果,最佳单倍体与参考序列片段进行SW比对后的第二SW比对结果;第一SW比对结果、第二SW比对结果由CIGAR表示,CIGAR由至少一个包含:系数和比对类型的操作连接而成,其中,比对类型可以包括:匹配/误配M、删除D、插入I中的至少一种或多种;
具体的,根据预设变换规则,对测序序列片段的SW比对结果进行转换,得到测序序列片段与参考序列片段的校正对齐结果,可以包括:
获取第二SW比对结果中的第一操作、以及第一SW比对结果中的第一操作,根据预设变换规则,对第二SW比对结果中的第一操作、以及第一SW比对结果中的第一操作进行比较,得到测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取新的一对Operator,根据预设变换规则对这一对新的Operator重新进行计算,待计算结束后,重新获取新的一对Operator,在根据预设变换规则重新计算,如此循环迭代计算得到测序序列片段与参考序列片段的校正对齐结果。
具体的,针对M、D、I三种比对类型,其具体比对时,该变化规则又进一步细化为M-M,M-D,M-I,D-M,D-D,D-I,I-M,I-D,I-I几对操作对应的变换规则,详见如下1)~9)所述:
获取第二SW比对结果中的第一操作、以及第一SW比对结果中的第一操作;其中,第二SW比对结果中的第一操作可以为第二SW比对结果中的任一操作,第一SW比对结果中的第一操作可以为第一SW比对结果中的任一操作。
1)若第二SW比对结果中的第一操作的比对类型为M,第一SW比对结果中的第一操作的比对类型为M,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第一SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、且比对类型为M的第二操作作为第二SW比对结果当前新的操作,将第二操作与下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果;并获取与第二SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为M的第三操作作为第一SW比对结果当前新的操作,将下一操作与第三操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
2)若第二SW比对结果中的第一操作的比对类型为M,第一SW比对结果中的第一操作的比对类型为D,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、比对类型为M的操作作为第二SW比对结果当前新的操作,将该新的操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将系数为第二SW比对结果中的第一操作的系数、比对类型为D的操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为D的操作作为第一SW比对结果当前新的操作,将下一操作与新的操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
3)若第二SW比对结果中的第一操作的比对类型为M,第一SW比对结果中的第一操作的比对类型为I,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将第二SW比对结果中的第一操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算。
4)若第二SW比对结果中的第一操作的比对类型为D,第一SW比对结果中的第一操作的比对类型为M,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将与第二SW比对结果中的第一操作相邻的下一操作与第一SW比对结果中的第一操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算。
5)若第二SW比对结果中的第一操作的比对类型为D,第一SW比对结果中的第一操作的比对类型为D,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将与第二SW比对结果中的第一操作相邻的下一操作与第一SW比对结果中的第一操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算。
6)若第二SW比对结果中的第一操作的比对类型为D,第一SW比对结果中的第一操作的比对类型为I,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将系数为第一SW比对结果中的第一操作的系数、比对类型为M的操作,以及系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、比对类型为D的操作顺序组合在一起作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将系数为第二SW比对结果中的第一操作的系数、比对类型为M的操作,以及系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为I的操作顺序组合在一起作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将系数为第二SW比对结果中的第一操作的系数、比对类型为M的操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
7)若第二SW比对结果中的第一操作的比对类型为I,第一SW比对结果中的第一操作的比对类型为M,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将系数为第一SW比对结果中的第一操作的系数、比对类型为I的操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、比对类型为I的操作作为第二SW比对结果当前新的操作,将该新的操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为M的操作作为第一SW比对结果当前新的操作,将下一操作与该新的操作与作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
8)若第二SW比对结果中的第一操作的比对类型为I,第一SW比对结果中的第一操作的比对类型为D,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则不输出测序序列片段与参考序列片段在当前位置的校正对齐结果,并生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、且比对类型为I的操作作为第二SW比对结果当前新的操作,将该新的操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则不输出测序序列片段与参考序列片段在当前位置的校正对齐结果,并将第二SW比对结果中的第一操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则不输出测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
9)若第二SW比对结果中的第一操作的比对类型为I,第一SW比对结果中的第一操作的比对类型为I,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将第二SW比对结果中的第一操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算。
如此,可以将测序序列片段、与测序序列片段比对最佳的单倍体、参考序列片段这3个序列相对于参考序列片段的第0个编码位置保持对齐的,每次比较该组SW比对结果中两个CIGAR的当前的比对类型,让测序序列片段以确保3个序列保持对齐的方式延伸合适的距离,依次不断循环比较,直至该组SW比对结果中两个CIGAR比较结束,测序序列片段也延伸至序列的最末尾,很好地实现了将测序序列片段与参考序列片段校正对齐。
在第一方面的再一种可实现方式中,结合第一方面或第一方面的任一种可实现方式,根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型可以包括:
根据校正比对结果,对潜在变异区域从前到后扫描每一个位置,确定潜在变异区域中的变异位置,判断每个变异位置中每种变异类型的测序序列片段的数量是否大于第三阈值,若存在M种变异类型,M种变异类型的测序序列片段的数量均大于第三阈值,则确定M种变异类型为基因组的变异类型。
如此,实现了根据测序序列片段的整体对齐结果,把具有相同变异类型的测序序列片段聚在一起对齐,避免将属于同一类型的变异错误地比对成不同类型的变异,从而提高基因组变异检测结果的准确性。
第二方面,本发明提供一种检测装置,用于执行第一方面所述的方法,该装置可以包括:
序列比对单元,用于将基因组的多条测序序列分别和参考序列进行双序列比对,得到双序列比对结果;
确定单元,用于根据序列比对单元得到的双序列比对结果,确定基因组的潜在变异区域;
创建单元,用于对每个潜在变异区域,在所有测序序列中抽取出处于潜在变异区域内的所有测序序列片段,根据所有测序序列片段创建迭代DeBruijn图,根据迭代DeBruijn图得到N个单倍体;其中,N为大于等于1的整数;
SW比对单元,用于对于潜在变异区域内的任一测序序列片段,将测序序列片段与N个单倍体进行SW比对,将测序序列片段的最佳单倍体与参考序列片段进行SW比对,得到一组与该测序序列片段对应的SW比对结果;
校正对齐单元,用于根据预设变换规则,对每个测序序列片段的SW比对结果进行转换,得到每个测序序列片段与参考序列片段的校正对齐结果;预设变换规则用于将每个测序序列片段与参考序列片段对齐;
确定单元,还用于根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型。
其中,第二方面的具体实现方式可以参考第一方面或第一方面的可实现方式提供的基因组变异检测方法中检测装置的行为功能,在此不再重复赘述。因此,第二方面提供的检测装置可以达到与第一方面相同的有益效果。
第三方面,本发明实施例提供一种检测装置,用于执行第一方面所述的方法,该装置可以包括:
处理器,用于将基因组的多条测序序列分别和参考序列进行双序列比对,得到双序列比对结果,根据序列比对单元得到的双序列比对结果,确定基因组的潜在变异区域,对每个潜在变异区域,在所有测序序列中抽取出处于潜在变异区域内的所有测序序列片段,根据所有测序序列片段创建迭代DeBruijn图,根据迭代DeBruijn图得到N个单倍体,对于潜在变异区域内的任一测序序列片段,将测序序列片段与N个单倍体进行SW比对,将测序序列片段的最佳单倍体与参考序列片段进行SW比对,得到一组与该测序序列片段对应的SW比对结果,根据预设变换规则,对每个测序序列片段的SW比对结果进行转换,得到每个测序序列片段与参考序列片段的校正对齐结果;预设变换规则用于将每个测序序列片段与参考序列片段对齐,根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型。
其中,第三方面的具体实现方式可以参考第一方面或第一方面的可实现方式提供的基因组变异检测方法中检测装置的行为功能,在此不再重复赘述。因此,第三方面提供的检测装置可以达到与第一方面相同的有益效果。此外,该设备还可以包括存储器,该存储器用于与处理器耦合,其保存该装置必要的程序指令和数据。
第四方面,本发明实施例提供一种存储一个或多个程序的非易失性计算机可读存储介质,该一个或多个程序包括指令,指令当被包括第二方面或第三方面或上述任一种可实现方式所述的检测装置执行时,使检测装置执行以下事件:
将基因组的多条测序序列分别和参考序列进行双序列比对,得到双序列比对结果,根据序列比对单元得到的双序列比对结果,确定基因组的潜在变异区域,对每个潜在变异区域,在所有测序序列中抽取出处于潜在变异区域内的所有测序序列片段,根据所有测序序列片段创建迭代DeBruijn图,根据迭代DeBruijn图得到N个单倍体,对于潜在变异区域内的任一测序序列片段,将测序序列片段与N个单倍体进行SW比对,将测序序列片段的最佳单倍体与参考序列片段进行SW比对,得到一组与该测序序列片段对应的SW比对结果,根据预设变换规则,对每个测序序列片段的SW比对结果进行转换,得到每个测序序列片段与参考序列片段的校正对齐结果;预设变换规则用于将每个测序序列片段与参考序列片段对齐,根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型。
其中,第四方面的具体实现方式可以参考第一方面或第一方面的可实现方式提供的基因组变异检测中检测装置的行为功能,在此不再重复赘述。因此,第四方面提供的检测装置可以达到与第三方面相同的有益效果。
附图说明
图1为本发明实施例提供的检测装置的结构图;
图2为本发明实施例提供的基因组变异检测方法的流程图;
图3为本发明实施例提供的确定潜在变异区域的过程示意图;
图3a为本发明实施例提供的一种基因组的编码区间划分示意图;
图3b为本发明实施例提供的一种测序序列片段和参考序列片段的抽取过程示意图;
图3c为本发明实施例提供的另一种测序序列片段和参考序列片段的抽取过程示意图;
图4为本发明实施例提供的创建迭代DeBruijn图的过程示意图;
图5a为本发明实施例提供的测序序列片段与单倍体进行SW比对的过程示意图;
图5b为本发明实施例提供的单倍体与参考序列片段进行SW比对的过程示意图;
图6为本发明实施例提供的测序序列片段与参考序列片段校正对齐的对齐结果示意图;
图7为本发明实施例提供的一种检测装置20的结构图。
具体实施方式
本发明实施例的基本原理是:使用局部组装创建迭代DeBruijn graph,遍历该迭代DeBruijn graph得到单倍体,然后借助预设变换规则实现的单倍体对齐的校正策略把覆盖每个变异位置的测序序列聚成不同的类,把具有相同变异类型的测序序列聚在一起对齐来确定基因组的变异类型,避免将属于同一类型的变异错误地比对成不同类型的变异,以此提高基因组变异检测结果的准确性。
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。详细描述本方案之前,为了便于理解本发明实施例所述的技术方案,对本发明实施例中的一些重要名词进行详细解释,需要理解的是,下述名词仅是本发明实施例技术人员为了描述方便进行的命名,并不代表或暗示所指的系统或元件必须有此命名,因此不能理解为对本发明实施例的限制:
基因组:包含人的所有遗传信息,而不单单只是一些外在和内在特征,也包含很多目前而言不明其功能性的碱基序列,基因是基因组的一个子集,基因是控制性状的遗传单元,性状为个体的各种外在和内在特征,比如:头发和眼睛颜色、高低胖瘦、抵抗力强等。在基因组的碱基序列中,按照碱基的排列顺序对每个碱基进行顺序编码,使每一个碱基具有一个相匹配的编码,单个编码代表基因组中的一个碱基对,连续的编码区间代表基因组中的一个碱基片段。
碱基序列:是一种由核苷酸分子构成的长链聚合物,这种长链聚合物具有方向性,上游末端称为5’端,下游末端称为3’端;其中,核苷酸分子也可称为碱基,不同的碱基可以相互组合,使得DNA序列形成双螺旋结构。碱基序列根据核苷酸分子上携带的四种不同碱基类型可以抽象为由字符集{A,C,G,T}构成的字符串,可以结合的碱基称为互补碱基,互补规则为A与T互补,G与C互补。
碱基对(英文:base pair,简称:bp):碱基序列长度的单位,是形成DNA单体以及编码遗传信息的化学结构。
测序(sequence):确定碱基序列内容的过程,如:将完整的样本碱基序列打碎,从中筛选出满足特定长度(通常为数百bp)的片段,然后在每个片段的一端或者两端各读取一段长度为数十至数百bp的序列,这些读取出的序列长度通常远远小于被测样本碱基序列的长度,使得全部短序列的总长度达到样本DNA长度的数倍至数十倍,从而使获得样本碱基序列成为可能。
供体(donor):在测序中提供被测序碱基序列的个体。
测序序列(read):是指一段特定长度的DNA片段,该DNA片段可以从打碎的碱基序列上读取出来。
局部封装(local assembly):是指将多个测序序列像拼图一样合并组装成更长的序列。通常情况下,可以寻找测序序列之间的重叠部分(overlap),从重叠部分将测序序列拼接起来,例如:下述两个测序序列:
ATACCTTGCTAGCGT
GCTAGCGTAGGTCTGA
经过局部组装之后成为:ATACCTTGCTAGCGTAGGTCTGA。
参考序列(reference sequence):“人类基因组计划”中所组装出来的人类基因组,是通过拼接方法获得的一套人类碱基序列。参考基因组并不是某个人类个体的基因组序列,它来自于多个具有代表性的人类种族个体的序列,是一套综合的序列。由于双链碱基序列呈互补关系,根据其中一条碱基序列即可以获得另一条序列内容,参考序列仅包括双链碱基序列中的一条。其他物种,如果蝇、小鼠等同样有参考序列,本发明实施例是针对人类基因组进行研究,故在本发明实施例中参考序列仅指人类基因组参考序列。其中,参考序列与碱基序列相同,都可以看作为定义在字符集{A,C,G,T}上的字符串,其中“A”、“C”、“G"、“T”分别对应核苷酸所携带的四种碱基。
基因组变异:是指基因组中碱基对组成或排列顺序的改变,主要包括:单核苷酸变异和indel(Insertion和Deletion的简称)两种;单核苷酸变异:通常称为单核苷酸多态性(英文:Single Nucleotide Polymorphism,SNP),通俗的说法就是单个DNA碱基的不同。indel:指的是基因组的某个位置上所发生的小片段序列的插入或删除,其长度通常在100bp以下。
kmer:是指将一条测序序列,连续切割挨个碱基滑动得到的一系列序列长度为k的碱基序列(即核苷酸序列),其中,k为kemr长度(size)。例如:测序序列为:
ATCGTTGCTTAATGACGTCAGTCGAATGCGATGACGTGACTGACTG
此时,若kmer size为13,则经过kmer后得到的一系列碱基序列为(需要说明的是,由于篇幅有限,在本发明实施例中,仅以kmer后得到的前四个碱基序列为例进行说明):
ATCGTTGCTTAAT
TCGTTGCTTAATG
CGTTGCTTAATGA
GTTGCTTAATGAC
………………………..
单倍型,是单倍体基因型的简称,在遗传学上是指在同一染色体上进行共同遗传的多个基因座上等位基因的组合;通俗的说法就是若干个决定同一性状的紧密连锁的基因构成的基因性。按照某一指定基因座上基因重组发生的数量,单倍型甚至可以指至少两个基因座或整个染色体。
本发明实施例提供的基因组变异检测方法可由图1所示的检测装置10执行,用于对基因组进行变异检测。如图1所示,所述检测装置10可以包括:通信单元1011、处理器1012、存储器1013以及至少一个通信总线1014,通信总线1014用于实现这些装置之间的连接和相互通信。
其中,通信单元1011可用于与外部网元或设备之间进行数据交互,如:可以收集样本碱基序列或者直接从基因数据库中读取样本碱基序列。或者,可以为一人机交互界面,用于将处理器1012检测后的处理反馈给检测人员。
处理器1012可能是一个中央处理器(英文:Central Processing Unit,CPU),也可以是特定集成电路(英文:Application Specific Integrated Circuit,ASIC),或者是被配置成实施本发明实施例的一个或多个集成电路,例如:一个或多个微处理器(英文:Digital Singnal Processor,DSP),或,一个或者多个现场可编程门阵列(英文:Field Programmable Gate Array,FPGA)。具体的,处理器1012可以用于确定被检测基因组中的潜在变异区域,对每个潜在变异区域,对落在该潜在变异区域内的测序序列进行局部组装得到迭代DeBruijn图,遍历迭代DeBruijn图的每条路径得到所有可能的haplotypes,把每个测序序列与所有可能的haplotypes中最佳的haplotype进行SW比对、以及把最佳的haplotype跟与潜在变异区域对应的参考序列进行SW比对,得到一组SW比对结果,根据该SW比对结果、以及预设的变换规则得到每个测序序列相对于潜在变异区域的参考序列校正对齐后的比对结果,根据每个测序序列校正对齐后的比对结果,确定基因变异类型。
存储器1013,可以是易失性存储器(英文:volatile memory),例如随机存取存储器(英文:Random-Access Memory,RAM);或者非易失性存储器(英文:non-volatile memory),例如只读存储器(英文:Read-Only Memory,ROM),快闪存储器(英文:flash memory),硬盘(英文:Hard Disk Drive,HDD)或固态硬盘(英文:Solid-State Drive,SSD);或者上述种类的存储器的组合。存储器1013可以用于存储数据和/或代码,处理器1012可以通过运行或执行存储在存储器1013内的程序代码,以及调用存储在存储器1013内的数据,实现基因组变异检测的功能。
通信总线1014可以分为地址总线、数据总线、控制总线等,可以是以太网总线、工业标准体系结构(英文:Industry Standard Architecture,ISA)总线、外部设备互连(英文:Peripheral Component,PCI)总线或扩展工业标准体系结构(英文:Extended Industry Standard Architecture,EISA)总线等。为便于表示,图1中仅用一条粗线表示各个通信总线,但并不表示仅有一根总线或一种类型的总线。
为了便于描述,以下实施例以步骤的形式示出并详细描述了本发明实施例中检测装置10执行的基因组变异检测方法,其中,示出的步骤也可以在除检测装置10之外的诸如一组可执行指令的计算机系统中执行。此外,虽然在图中示出了逻辑顺序,但是在某些情况下,可以以不同于此处的顺序执行所示出或描述的步骤。
图2为本发明实施例提供的基因组变异检测方法的流程图,由图1所示的检测装置10执行,如图2所示,所述方法可以包括以下步骤:
步骤101:将基因组的多条测序序列分别和参考序列进行双序列比对,得到双序列比对结果。
其中,步骤101中的基因组为:被检测基因组,该被检测基因组可以为待通过本发明实施例提供的检测方法检测出基因组变异类型的基因组。可选的,该基因组可以由本发明实施例提供的检测装置直接从供体上获取,或者由检测装置从存储有大量基因组的基因组数据库中获取,本发明实施例对此不进行限定。
参考序列为基因组没有发生变异时的碱基序列,其代表了基因组中碱基正确的排列顺序,因此,可以以参考序列为基准判断测序序列的变异情况,当测序序列与参考序列的碱基排列顺序一致时,说明测序序列没有发生变异;当测序序列与参考序列的碱基排列顺序不一致时,说明测序序列发生了变异,其中,测序序列的变异类型主要包括碱基的错配、插入和删除。
测序序列为短序列片段,可选的,可以通过高通量测序仪获取基因组的多个测序序列;需要说明的是,测序序列的数量越多,在基因组变异检测过程中得到的原始数据越多,在后续步骤中对基因组变异检测结果进行统计分析时,可利用的数据越多,则基因组变异检测结果越准确,因此,步骤101中测序序列的个数要尽可能的多。
双序列比对结果可以包含:每条测序序列相对参考序列详细的变异信息,如:可以包括匹配(match)、错配(mistake)、插入(insertion)或删除(deletion)等信息。其中,双序列比对结果可以用文本格式(如SAM(英文:Sequence Alignment/Map Format)格式)表示,还可以用二进制压缩格式(如BAM(英文:Binary Alignment/Map Format)格式)表示,本发明实施例对此不进行限定。
可选的,可以通过BWA等短序列对比软件将基因组的多条测序序列分别与基因组的参考序列进行双序列比对,即将每条测序序列定位到参考序列的相应位置,获得每条测序序列相对参考序列详细的变异信息。
步骤102:根据双序列比对结果,确定基因组的潜在变异区域。
其中,基因组的潜在变异区域可以指:基因组中很有可能存在变异的编码区间。
可选的,在本发明实施例中,可以通过下述1)、2)任一种方式来确定基因组的潜在变异区域:
1)采用基因分析工具(英文:The Genome Analysis Toolkit,GATK)中子模块HaplotypeCaller基于熵的过滤策略来根据双序列比对结果,确定基因组的潜在变异区域,其具体实现可以如图3所示,包括以下步骤:
1011:根据基因组的编码顺序,将基因组划分为多个编码区间。
其中,在将基因组划分为多个编码区间的过程中,编码区间的长度可以根据实际需要相应调整,例如,可以选择50-300bp范围内的任一长度作为一个编码区间,本发明实施例对此不做限制。需要说明的是,由于参考序列为基因组没有发生变异时的碱基序列,因此,在实际应用中可以用参考序列的编码区间代表基因组的编码区间。
1012:根据双序列比对结果,确定所有测序序列的变异类型。
由于测序序列和参考序列的双序列比对结果中包括测序序列相对于参考序列详细的匹配、错配、插入和删除等信息,因此,可以根据双序列比对结果可以直接确定测序序列的变异类型。
需要说明的是,在本发明实施例中,相同变异类型的测序序列是指相对参考序列具有完全相同变异信息的测序序列,没有发生变异的测序序列也作为变异类型的一种。
1013:统计每个编码区间内不同变异类型的测序序列的概率分布值。
其中,编码区间内每种变异类型的测序序列的概率分布值为:编码区间内该类变异类型的测序序列的数量与编码区间内总的测序序列总数的比值,该概率分布值可以记为pi。
假如在编码区间内存在两种变异类型,分别为第一变异类型和第二变异类型,分别统计第一变异类型和第二变异类型的测序序列的数量,将第一变异类型的数量除以测序序列总数,得到第一变异类型的概率p1,将第二变异类型的数量除以测序序列总数,得到第二变异类型的概率p2,p1和p2即编码区间内不同变异类型的测序序列的概率分布值。
1014:根据编码区间内不同变异类型的测序序列的概率分布值,计算编码区间的信息熵。
其中,信息熵为本领域通用概念,可以用于反映序列的混杂程度,信息熵越大说明序列越混乱,测序序列发生变异的可能性就越大,因此,本发明实施例可以通过信息熵确定潜在变异区域。
可选的,可以将编码区间内不同变异类型的测序序列的概率分布值pi代入信息熵公式:H(U)=E[-logpi],得到编码区间的信息熵H(U)。
1015:判断每个编码区间的信息熵是否大于第一阈值,若存在第一编码区间,其信息熵大于第一阈值,则将该第一编码区间确定为潜在变异区域。
需要指出的是,在一个基因组中,潜在变异区域的数量可以为一个或一个以上的多个,本发明对此不做限制。
其中,第一阈值可以根据需要进行设置,本发明实施例对此不进行限定。当编码区间的信息熵大于第一阈值,则表示编码区间内序列越混乱,编码区间为潜在变异区域的可能性就越大。
例如:图3a为本发明实施例提供的一种基因组的编码区间划分示意图,由于参考序列为基因组没有发生变异时的碱基序列,因此,在图3a中可以用参考序列的编码区间代表基因组的编码区间。如图3a所示,在本实施例中,沿着基因组的编码顺序,将基因组划分为长度为100bp的编码区间,依次形成第一编码区间(1510531,1510630)、第二编码区间(1510631,1510730)、第三编码区间(1510731,1510830)、第四编码区间(1510831,1510930)等,在编码区间划分完成后,可以采用上述方式依次判断每个编码区间是否为潜在变异区域,在所有编码区间中筛选出基因组的潜在变异区域。
2)采用其他的自定义的启发式策略确定基因组的潜在变异区域,其具体实现可以包括以下步骤:
根据基因组的编码顺序,将基因组划分为多个编码区间;
依次统计每个编码区间内发生变异的测序序列的数量;
判断每个编码区间内发生变异的测序序列的数量是否大于第二阈值;
若存在第一编码区间,其内发生变异的测序序列的数量大于第二阈值,则确定该第一编码区间为潜在变异区域。
其中,方式2)中划分多个编码区间的过程可以与方式1)中划分多个编码区间的过程相同,在此不再重复赘述。
发生变异的测序序列可以指:测序序列和参考序列不能完美匹配的测序序列,如:可以为存在错配、插入或删除的测序序列。
第二阈值可以根据需要进行设置,本发明实施例对此不进行限定。当发生变异的测序序列的数量大于第二阈值,则表示编码区间内发生变异的测序序列的数量较多,编码区间为潜在变异区域的可能性就越大。
例如,将第二阈值设定为50,则当发生变异的测序序列的数量大于50时,确定所述编码区间为潜在变异区域,否则,不将所述编码区域间作为潜在变异区域。
步骤103:在所有测序序列中抽取出处于所述潜在变异区域内的所有测序序列片段,根据所有测序序列片段创建迭代DeBruijn图,根据迭代DeBruijn图得到N个单倍体。
其中,N可以为大于等于1的整数。
可选的,在本发明实施例的一种可能的实现方式中,抽取出处于所述潜在变异区域内的测序序列片段可以包括:
抽取每条测序序列与所述潜在变异区域的交集部分作为测序序列片段;例如,当潜在变异区域的编码区间与测序序列的编码区间的交集为测序序列的编码区间时,抽取整条所述测序序列作为测序序列片段;当潜在变异区域的编码区间与测序序列的编码区间的交集为测序序列的编码区间的一部分时,抽取所述测序序列与潜在变异区域的交集部分作为测序序列片段;当潜在变异区域的编码区间与测序序列的编码区间的交集为空集时,将所述测序序列丢弃。
图3b为本发明实施例提供的一种测序序列片段和参考序列片段的抽取过程示意图,在图3b中,以三种不同类型的测序序列为例,对测序序列片段的抽取过程进行示例性说明。其中,潜在变异区域的编码区间为(1510531,1510630),第一测序序列的编码区间为(1510541,1510590),第二测序序列的编码区间为(1510521,1510570),第三测序序列的编码区间为(1510651,15106700)。
对于第一测序序列,其编码区间(1510541,1510590)完全处于潜在变异区域的编码区间(1510531,1510630)内,则抽取完整的第一测序序列作为测序序列片段;对于第二测序序列,其编码区间(1510521,1510570)与潜在变异区域的编码区间(1510531,1510630)存在部分交集(1510531,1510570),则在第二测序序列中抽取编码区间(1510531,1510570)部分作为测序序列片段;对于第三测序序列,其编码区间(1510651,15106700)与潜在变异区域的编码区间(1510531,1510630)的交集为空集,则将第三测序序列丢弃,从而抽取到的测序序列片段为第一测序序列的全部以及第二测序序列编码区间为(1510531,1510570)的部分。
或者,在本发明实施例的另一种可能的实现方式中,抽取出处于所述潜在变异区域内的测序序列片段可以包括:
判断每条测序序列与所述潜在变异区域是否存在交集;当测序序列与所述潜在变异区域存在交集时,抽取所述测序序列作为测序序列片段。即当潜在变异区域的编码区间与测序序列的编码区间的交集为测序序列的编码区间的一部分时,以测序序列的编码区间为基准对潜在变异区域进行扩展,避免在测序序列片段的抽取过程中将测序序列打断,保证测序序列的完整性。
例如:图3c为本发明实施例提供的另一种测序序列片段和参考序列片段的抽取过程示意图,在图3c中测序序列片段的抽取过程与图3b基本相似,其不同之处在于,对于第二测序序列,由于其编码区间与潜在变异区域的编码区间存在部分交集,则将第二测序序列的编码区间(1510521,1510570)与潜在变异区域的编码区间(1510531,1510630)的并集(1510521,1510630)作为扩展后的潜在变异区域的编码区间,然后在第二测序序列的潜在变异区域内抽取测序序列片段。由于第二测序序列的编码区间完全落在扩展后的潜在变异区域的编码区间内,因此,抽取整条第二测序序列作为测序序列片段。
迭代DeBruijn图的概念源自《IDBA-A Practical Iterative DeBruijn Graph De Novo Assembler》这篇文章(paper),作者通过把组装的kmer size从最小kmer size(kmin)迭代到最大kmer size(kmax)来提高全基因组组装(denovo assembly)的精度,所以提出了迭代DeBruijn图这个非常形象的概念。但是全基因组denovo assembly与用于变异检测的local assembly在构建DeBruijn graph时还是存在着非常明显的差异:全基因组denovo assembly的目标是装出尽可能长的Contig,因此通常会合并图上的一些SNP/indel造成的分叉而去掉SNP/indel(即通过牺牲局部区域的组装精度来获得更长的组装Contig);而用于变异检测的local assembly的目标则是尽量准确无遗漏地检测到各种SNP/indel变异,所以通常有必要保留DeBruijn图上代表的SNP/indel的小分叉。因此,针对变异检测的local assembly的需求,在本发明实施例中,可选的,可以采取如图4所示步骤来创建迭代DeBruijn图:
设定初始kmer size,即最小kmer size(kmin)以及最大kmer size(kmax)。
步骤2011:根据初始kmer size的值kmin,从所有测序序列当中过滤掉支持数低于设定阈值的kmer,并以kmer作为节点,过滤后的测序序列上的相邻kmer作为边,创建最初始的DeBruijn图。
步骤2022:遍历当前DeBruijn图产生多个contig,过滤出长度大于测序仪读长的contig作为新扩充的测序序列,并删除掉测序序列中被所述新扩充的测序序列完全包含的测序序列,同时,将k值加1。
步骤2023:以所有测序序列中剩下的测序序列和所述新扩充的测序序列按照新的k值,以所述kmer作为节点,测序序列上的相邻kmer作为边创建新的DeBruijn图;其中,所述k值为kmer长度,所述当前DeBruijn图根据所述k值创建。
重复步骤上述步骤2022~2023,直至kmer size为预设的最大kmer size(kmax),此时的DeBruijn图即为最终所需要的迭代DeBruijn图。
其中,最小kmer size(kmin)以及最大kmer size(kmax)可以根据需要设置,本发明实施例对此不进行限定。
可选的,根据迭代DeBruijn图得到N个单倍体可以包括:
对迭代DeBruijn图中权重支持数小于预设权重的连接边进行修剪;
遍历修剪后的迭代DeBruijn图中的每条路径,得到N个单倍体。
步骤104:对于潜在变异区域内的任一测序序列片段,将该测序序列片段与N个单倍体进行SW比对,将该测序序列片段对应的最佳单倍体与参考序列片段进行SW比对,得到一组与该测序序列片段对应的SW比对结果。
其中,该测序序列片段对应的SW比对结果包括:该测序序列片段与最佳单倍体进行SW比对后的第一SW比对结果,最佳单倍体与参考序列片段进行SW比对后的第二SW比对结果。
参考序列片段为:在参考序列中处于潜在变异区域内的参考序列片段;可选的,可以以潜在变异区域的编码区间为基准,在参考序列中抽取出参考序列片段。例如,在图3b中,潜在变异区域的编码区间为(1510531,1510630),则在参考序列中抽取出编码区间(1510531,1510630)部分,作为参考序列片段;在图3c中,扩展后的潜在变异区域的编码区间为(1510521,1510630),则在参考序列中抽取出编码区间(1510521,1510630)部分,作为参考序列片段。
最佳单倍体为:N个单倍体中与测序序列片段对比结果最佳的单倍体,可选的,可以将该测序序列片段与N个单倍体中每个单倍体进行SW比对),将对比后得分最高所对应的单倍体确定为与该测序序列片段对应的最佳单倍体,其中,SW比对算法为生物信息学领域的通用算法,在此不再详细赘述。
在本发明实施例中,SW对比结果可以采用CIGAR的形式来表示,其中,CIGAR是一个字符串,其基本形式为系数和一个字符连接形成的操作,可以由多个操作连接形成,系数用来表示序列的长度,字符用来表示该段序列的比对类型,常用的比对类型包括:M、D、I;“M”,表示测序序列片段与参考序列片段匹配或者存在替换(即Match/MisMatch);“D”,表示参考序列片段与测序序列片段的空位相对应,即deletion;“I”,表示测序序列片段与参考序列片段的空位相对应,即insertion。
例如,如图5a所示,为测序序列片段与单倍体的SW比对结果,写成CIGAR表示就是38D69M47D,即测序序列片段相对于单倍体删除了单倍体的前38个碱基、又跟单倍体接下来的69个碱基匹配上了、最后又删除了单倍体接下来的47个碱基;如图5b所示,为单倍体与参考序列片段的SW比对结果,写成CIGAR表示就是59M2D16M1I78M,即单倍体先与测序序列片段的59个碱基匹配上了、接下来单倍体相对于参考序列片段删除了参考序列片段的2个碱基、又跟参考序列片段接下来的16个碱基匹配上了、接下来单倍体相对于参考序列片段插入了1个碱基、最后又跟参考序列片段的78个碱基匹配上了。
步骤105:根据预设变换规则,对每个测序序列片段的SW比对结果进行转换,得到每个测序序列片段与参考序列片段的校正对齐结果。
其中,校正对齐结果包含:将测序序列片段与参考序列片段校正对齐后,测序序列片段详细的变异信息,如:包括测序序列片段的变异位置和变异位置处错配、插入或删除信息。
预设变换规则用于将测序序列片段与参考序列片段校正对齐;其核心思想是:将测序序列片段、与测序序列片段比对最佳的单倍体、参考序列片段这3个序列相对于参考序列片段的第0个编码位置保持对齐的,每次比较该组SW比对结果中两个CIGAR的当前的比对类型,让测序序列片段以确保3个序列保持对齐的方式延伸合适的距离,依次不断循环比较,直至该组SW比对结果中两个CIGAR比较结束,测序序列片段也延伸至序列的最末尾。
具体的,由于比对类型包括:M、D、I,因此,具体比对时,该规则又进一步细化为M-M,M-D,M-I,D-M,D-D,D-I,I-M,I-D,I-I9等几种组合对应的变换规则,其中,每一种组合,第一个操作(Operator)来自于测序序列片段对应的比对最佳的单倍体haplotype相对于参考序列的CIGAR,第二个Operator来自于测序序列片段相对于其比对最佳的单倍体的CIGAR,对每组组合,其比对规则如下所示:
获取第二SW比对结果中的第一操作、以及第一SW比对结果中的第一操作;其中,第二SW比对结果中的第一操作可以为第二SW比对结果中的任一操作,第一SW比对结果中的第一操作可以为第一SW比对结果中的任一操作。
1)若第二SW比对结果中的第一操作的比对类型为M,第一SW比对结果中的第一操作的比对类型为M,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第一SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、且比对类型为M的第二操作作为第二SW比对结果当前新的操作,将第二操作与下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果;并获取与第二SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为M的第三操作作为第一SW比对结果当前新的操作,将下一操作与第三操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
2)若第二SW比对结果中的第一操作的比对类型为M,第一SW比对结果中的第一操作的比对类型为D,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、比对类型为M的操作作为第二SW比对结果当前新的操作,将该新的操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将系数为第二SW比对结果中的第一操作的系数、比对类型为D的操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为D的操作作为第一SW比对结果当前新的操作,将下一操作与新的操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
3)若第二SW比对结果中的第一操作的比对类型为M,第一SW比对结果中的第一操作的比对类型为I,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将第二SW比对结果中的第一操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算。
4)若第二SW比对结果中的第一操作的比对类型为D,第一SW比对结果中的第一操作的比对类型为M,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将与第二SW比对结果中的第一操作相邻的下一操作与第一SW比对结果中的第一操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算。
5)若第二SW比对结果中的第一操作的比对类型为D,第一SW比对结果中的第一操作的比对类型为D,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将与第二SW比对结果中的第一操作相邻的下一操作与第一SW比对结果中的第一操作作为新的一对操作Operator,并根据预设变换规则对这一对新的Operator进行计算。
6)若第二SW比对结果中的第一操作的比对类型为D,第一SW比对结果中的第一操作的比对类型为I,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将系数为第一SW比对结果中的第一操作的系数、比对类型为M的操作,以及系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、比对类型为D的操作顺序组合在一起作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将系数为第二SW比对结果中的第一操作的系数、比对类型为M的操作,以及系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为I的操作顺序组合在一起作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将系数为第二SW比对结果中的第一操作的系数、比对类型为M的操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
7)若第二SW比对结果中的第一操作的比对类型为I,第一SW比对结果中的第一操作的比对类型为M,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则将系数为第一SW比对结果中的第一操作的系数、比对类型为I的操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、比对类型为I的操作作为第二SW比对结果当前新的操作,将该新的操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二SW比对结果中的第一操作相邻的下一操作、以及生成新的系数为第一SW比对结果中的第一操作的系数与第二SW比对结果中的第一操作的系数的差、比对类型为M的操作作为第一SW比对结果当前新的操作,将下一操作与该新的操作与作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则将第二SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
8)若第二SW比对结果中的第一操作的比对类型为I,第一SW比对结果中的第一操作的比对类型为D,则比较第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数大,则不输出测序序列片段与参考序列片段在当前位置的校正对齐结果,并生成新的系数为第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数的差、且比对类型为I的操作作为第二SW比对结果当前新的操作,将该新的操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数比第一SW比对结果中的第一操作的系数小,则不输出测序序列片段与参考序列片段在当前位置的校正对齐结果,并将第二SW比对结果中的第一操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算;
若第二SW比对结果中的第一操作的系数与第一SW比对结果中的第一操作的系数相等,则不输出测序序列片段与参考序列片段在当前位置的校正对齐结果,并获取与第二比对结果SW中的第一操作相邻的下一操作、以及获取与第一SW比对结果中的第一操作相邻的下一操作,根据预设变换规则对这两个下一操作进行计算。
9)若第二SW比对结果中的第一操作的比对类型为I,第一SW比对结果中的第一操作的比对类型为I,则将第一SW比对结果中的第一操作作为测序序列片段与参考序列片段在当前位置的校正对齐结果,并将第二SW比对结果中的第一操作与第一SW比对结果中的第一操作相邻的下一操作作为新的一对Operator,并根据预设变换规则对这一对新的Operator进行计算。
为了使上述变换规则更加明确、清楚,下面通过a)~i)来说明上述9种组合对应的变换规则:
a)M-M,此时要比较两个M系数的大小,若前者大,例如6M-4M,则先得到4M,再把2M跟4M的下一个Operator进行比较;若后者大,例如4M-6M,则先得到4M,再把4M的下一个Operator跟2M进行比较;若两者系数相等,例如2M-2M,则直接得到2M,然后各取下一个Operator进行比较。
b)M-D,此时要比较M和D系数的大小,若前者大,例如3M-2D,则先由2M-2D得到2D,再把1M跟2D的下一个Operator进行比较;若后者大,例如2M-3D,则先让2M-2D得到2D,再把2M的下一个Operator跟1D进行比较;若两者系数相等,例如2M-2D,则直接得到2D,然后各取下一个Operator进行比较。
c)M-I,此时M和I系数的大小没有影响,例如若为3M-2I,则先得到2I,再把3M跟2I的下一个Operator进行比较。
d)D-M,此时D和M系数的大小没有影响,例如若为3D-2M,则先得到3D,再把3D的下一个Operator跟2M进行比较。
e)D-D,此时两个D系数的大小没有影响,例如若为2D-5D,则先得到2D,再把2D的下一个Operator跟5D进行比较。
f)D-I,此时要比较D和I系数的大小,若后者大,例如2D-3I,则先得到2M1I,然后各取下一个Operator进行比较;若为前者大,例如3D-2I,则先得到2M1D,然后各取下一个Operator进行比较;若两者系数相等,例如2D-2I,则先得到2M,然后各取下一个Operator进行比较。
g)I-M,此时要比较I和M系数的大小,若前者大,例如5I-2M,则先由2I-2M得到2I,再把3I跟2M的下一个Operator进行比较;若后者大,例如2I-5M,则先由2I-2M得到2I,再把2I的下一个Operator跟3M进行比较;若两者系数相等,例如2I-2M,则先由2I-2M得到2I,然后各取下一个Operator进行比较。
h)I-D,此时要比较I和D系数的大小,若前者大,例如5I-2D,则先2I-2D直接消除,再把3I跟2D的下一个Operator进行比较;若后者大,例如2I-5D,则先2I-2D直接消除,再把2I的下一个Operator跟3D进行比较;若两者系数相等,例如2I-2D,则先2I-2D直接消除,然后各取下一个Operator进行比较。
i)I-I,此时两个I系数的大小没有影响,例如若为2I-5I,则先得到5I,再把2I跟5I的下一个Operator进行比较。
例如:若haplotype相对于参考序列的CIGAR为:59M2D16M1I78M,
read相对于haplotype的CIGAR为:38D69M47D,
则按照上述变换规则进行变换的过程如下所示:
59M-38D,前者的系数比后者大,则先得到38D,再把21M跟38D的下一个操作69M进行比较;
21M-69M,后者的系数比前者大,则先得到21M,再把59M的下一个操作2D跟48M进行比较;
2D-48M,后者的系数比前者大,则先得到2D,再把2D的下一个操作16M跟48M进行比较;
16M-48M,后者的系数比前者大,则先得到16M,再把16M的下一个操作1I跟32M进行比较;
1I-32M,后者的系数比前者大,则先得到1I,再把1I的下一个操作78M跟31M进行比较;
78M-31M,先得到31M,再把47M跟47D进行比较;
47M-47D,得到47D,两个CIGAR的Operator比较结束;
所以按照规则的最终计算结果为38D21M2D16M1I31M47D。
另外,在极少数情况下,可能会出现两个CIGAR其中一个CIGAR的所有Operator已经提前比较结束了,此时只需要直接拷贝未结束的CIGAR剩下的Operator即可。
步骤106:根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型。
由于校正对齐结果中具有测序序列片段与参考序列片段对齐后的变异信息,因此,根据校正对齐结果可以将多个测序序列片段对齐到参考序列片段上,并且可以根据校正结果中的变异信息确定潜在变异区域中的变异位置。
可选的,在本发明实施例中,根据每个测序序列片段与参考序列片段的校正对齐结果,确定基因组的变异类型可以包括:
根据校正比对结果,对潜在变异区域从前到后扫描每一个位置,确定潜在变异区域中的变异位置;
若当前位置存在变异则根据变异类型,统计每种变异类型的测序序列片段的数量;
判断每个变异位置中每种变异类型的测序序列片段的数量是否大于第三阈值,若存在M种变异类型,所述M种变异类型的测序序列片段的数量均大于第三阈值,则确定所述M种变异类型为所述基因组的变异类型;其中,所述M为大于等于1的整数。
例如,在图6中,根据测序序列片段与第一参考序列片段的校正对齐结果,确定编码1510581处为潜在变异区域中的变异位置;提取所有测序序列片段在编码1510581处的变异信息,共存在三种,分别为:不存在变异,存在碱基段CCT插入,存在碱基段CCT删除;根据所述变异信息,将所有测序序列片段汇聚至三个测序序列簇,分别为第一测序序列簇(变异信息为不存在变异,测序序列片段的数量为11条)、第二测序序列簇(变异信息为存在碱基段CCT插入,测序序列片段的数量为7条)和第三测序序列簇(变异信息为存在碱基段CCT删除,测序序列片段的数量为8条);依次判断每个测序序列簇中的测序序列片段的数量是否大于第三阈值。
假如第三阈值为6,则上述三个测序序列簇中测序序列片段的数量均大于第三阈值,从而得到基因组在编码1510581处的变异类型为:不存在变异;碱基段CCT插入;碱基段CCT删除。
假如第三阈值为10,则在上述三个测序序列簇中只有第一测序序列簇中测序序列片段的数量大于第三阈值,从而得到基因组在编码1510581处的变异检测结果为:不存在变异。
需要指出的是,上述第三阈值的大小仅是本发明实施例中的示例性说明,本领域的技术人员可以根据实际需要对第三阈值的大小进行相应调整,其均应当落入本发明的保护范围之内。
需要说明的是,图2所示过程主要对一个潜在变异区域中的变异类型检测,对于基因组中的其他潜在变异类型,可以采用图2所示的方案进行检测,在此不再重复赘述。
由上可知,与现有技术相比,图2所示的技术方案只需创建单张迭代DeBruijn图,减少了DeBruijn图的创建个数,进而减少单倍体的个数,使得减少单倍体与参考序列进行SW比对的次数大大降低,提升了比对速度;其次,图2所示的技术方案中通过先把每个测序序列片段定位到其比对最佳的单倍体上,然后借助该单倍体与参考序列片段间的比对结果,根据预设转换规则,把该测序序列片段校正对齐到参考序列上,实现了测序序列片段的整体对齐,便于把具有相同变异类型的测序序列片段聚在一起对齐,避免将属于同一类型的变异错误地比对成不同类型的变异,从而提高基因组变异检测结果的准确性。
上述主要从检测装置的角度对本发明实施例提供的基因组变异检测方法进行了介绍。可以理解的是,上述设备为了实现上述功能,其包括了执行各个功能相应的硬件结构和/或软件模块。本领域技术人员应该很容易意识到,结合本文中所公开的实施例描述的各示例的单元及算法步骤,本发明能够以硬件或硬件和计算机软件的结合形式来实现。某个功能究竟以硬件还是计算机软件驱动硬件的方式来执行,取决于技术方案的特定应用和设计约束条件。专业技术人员可以对每个特定的应用来使用不同方法来实现所描述的功能,但是这种实现不应认为超出本发明的范围。
本发明实施例可以根据上述方法示例、结合附图7对检测装置进行功能模块的划分,例如,可以对应各个功能划分各个功能模块,也可以将两个或两个以上的功能集成在一个处理模块中。上述集成的模块既可以采用硬件的形式实现,也可以采用软件功能模块的形式实现。需要说明的是,本发明实施例中对模块的划分是示意性的,仅仅为一种逻辑功能划分,实际实现时可以有另外的划分方式。
在采用对应各个功能划分各个功能模块的情况下,图7为本发明实施例中所涉及的检测装置20的一种可能的结构示意图,如图7所示,检测装置20可以用于实施上述方法实施例中检测装置所执行的方法,该检测装置20可以包括:序列比对单元201、确定单元202、创建单元203、SW比对单元204、校正对齐单元205;其中,序列比对单元101用于支持检测装置执行图2所示的步骤101和步骤106,确定单元202用于支持检测装置执行图2所示的步骤102,创建单元203用于支持检测装置执行图2所示的步骤103,SW比对单元204用于支持检测装置执行图2所示的步骤104,校正对齐单元205用于支持检测装置执行图2所示的步骤105。
需要说明的是,上述方法实施例涉及的各步骤的所有相关内容均可以援引到对应功能模块的功能描述,在此不再重复赘述。
在采用集成的单元的情况下,图7所示的序列比对单元201、确定单元202、创建单元203、SW比对单元204、校正对齐单元205可以集成在一个或者多个处理模块中,该处理模块可以为图1所示的检测装置的处理器1012,由处理器1012执行上述序列比对单元201、确定单元202、创建单元203、SW比对单元204、校正对齐单元205的功能。进一步的,检测装置20还可以包括存储模块和通信模块,存储模块用于存储检测装置20的程序代码和数据,处理模块用于执行存储模块中的程序代码和数据以实现上述方法实施例中转发节点的相应功能,存储模块可以为图1所示的存储器1013,通信模块可以为图1所示的通信单元1011。
所属领域的技术人员可以清楚地了解到,为描述的方便和简洁,上述描述的单元和系统的具体工作过程,可以参考前述方法实施例中的对应过程,在此不再赘述。
在本申请所提供的几个实施例中,应该理解到,所揭露的系统,设备和方法,可以通过其它的方式实现。例如,以上所描述的设备实施例仅仅是示意性的,例如,所述单元的划分,仅仅为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。
所述作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个网络单元上。可以根据实际的需要选择其中的部分或者全部单元来实现本实施例方案的目的。
另外,在本发明各个实施例中的各功能单元可以集成在一个处理单元中,也可以是各个单元单独物理,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用硬件加软件功能单元的形式实现。
上述以软件功能单元的形式实现的集成的单元,可以存储在一个计算机可读取存储介质中。上述软件功能单元存储在一个存储介质中,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行本发明各个实施例所述方法的部分步骤。而前述的存储介质包括:U盘、移动硬盘、只读存储器(Read-Only Memory,简称ROM)、随机存取存储器(Random Access Memory,简称RAM)、磁碟或者光盘等各种可以存储程序代码的介质。
本领域普通技术人员可以理解上述实施例的各种方法中的全部或部分步骤是可以通过程序来指令相关的硬件(例如处理器)来完成,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘或光盘等。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!