用于发现预测对检查点抑制剂敏感的MSI和新表位的系统、组合物和方法与流程
技术领域
本发明的领域是用于预测治疗选择的组学数据的计算分析,尤其涉及预测肿瘤对一种或多种检查点抑制剂的阳性治疗反应。
背景技术:
背景描述包括对理解本发明可能有用的信息。其并不是承认本文提供的任何信息是现有技术或与当前要求保护的发明相关,或者明确或暗示引用的任何出版物是现有技术。
本文中的所有出版物和专利申请通过引用并入本文,就如同每个单独的出版物或专利申请被具体地和单独地指出通过引用并入。当并入的参考文献中的术语的定义或使用与本文提供的术语的定义不一致或相反时,本文提供的该术语的定义适用并且该参考文献中对该术语的定义不适用。
癌症免疫疗法已经在一些患者中引起显著的反应,但是,尽管与免疫疗法应答者相比具有明显相同类型的癌症,但许多患者未能产生应答。这种失败的一种可能的解释是免疫系统的各种效应细胞可以被与一种或多种抑制性调控途径相互作用的化合物(检查点抑制剂)阻断。值得注意的是,一些肿瘤细胞能够利用抑制性调控途径来逃避免疫系统的检测和破坏。除了其他组分,PD-1和CTLA-4是与抑制免疫应答有关的受研究最多的受体,并且现在阻断这些受体活化的特异性药物已经变得可用。例如,针对PD-1(例如,纳武单抗(nivolumab)和派姆单抗(pembrolizumab))和CTLA4(例如易普利姆玛(ipilimumab))的抗体已经在黑素瘤、肾细胞癌、非小细胞肺癌和各种其他肿瘤类型的一些病例中产生显著的临床反应。不幸的是,并非所有类型的癌症都对检查点抑制剂的治疗反应良好。此外,即使在相同类型的癌症内,检查点抑制剂的阳性反应可预测性也是难以捉摸的。
另外,失配修复(MMR)的丧失通常通过损害细胞检测DNA损伤并激活细胞凋亡的能力直接导致耐药性,并且通过增加整个基因组中的突变率来间接导致耐药性。例如,已报道MMR缺陷型细胞对各种甲基化/烷基化剂、某些含铂药物、抗代谢物和拓扑异构酶II抑制剂具有抗性。此外,MMR缺陷细胞具有增加的突变率,其通常表示为微卫星不稳定性(MSI)。由于这些细胞通常对常规药物治疗较不敏感,因此免疫治疗是可取的。然而,免疫治疗对MSI肿瘤的疗效正如用检查点抑制剂治疗MSI肿瘤一样是不可预测的。
因此,希望有一种预测工具可以帮助评估用检查点抑制剂单独或联合靶向患者特异性和癌症特异性新表位治疗的癌症治疗功效
技术实现要素:
本发明主题涉及用于预测肿瘤对检查点抑制剂的治疗反应的各种装置、系统和方法。在特别优选的方面,然后使用HLA匹配的新表位的存在和数量(和模式)作为检查点抑制剂可能治疗成功的替代性指标。
在本发明主题的一个方面,本发明人考虑了一种使用免疫疗法来改善癌症治疗的方法,其包括从患者获得肿瘤组织以及匹配的正常组织的组学数据步骤,以及使用组学数据以确定多个基于错义的患者特异性和肿瘤特异性新表位的另一个步骤。在进一步的步骤中,然后过滤并定量新表位以获得HLA匹配的新表位。然后,当确定HLA匹配的新表位的量已超过预定阈值量时,将检查点抑制剂(例如CTLA-4抑制剂或PD-1抑制剂)施用于患者。
更典型地,使用多个不同的个体新表位序列(例如各自长度为7至20个氨基酸)对新表位中的每一个进行过滤新表位的步骤,其中改变的氨基酸在新表位序列内具有不同的位置。还考虑了过滤步骤可进一步包括通过事先已知的分子变异进行过滤的步骤,所述分子变异例如单核苷酸多态性、短缺失和插入多态性、微卫星标志物、短串联重复、杂合序列、多核苷酸多态性或命名变体。在进一步考虑的方面,过滤步骤还可包括测定新表位与该患者的至少一种I型MHC亚型和/或至少一种II型MHC亚型的亲和力,并还可包括测定新表位的表达水平。
尽管不限于本发明主题,但考虑了HLA匹配的新表位将对患者的至少一种I型MHC亚型和/或至少一种II类MHC亚型具有等于或小于150nM的亲和力。例如,考虑了定量HLA匹配的新表位的步骤可包括定量新表位对患者的至少一个I型MHC亚型或至少一种II类MHC亚型的亲和力(例如等于或小于500nM,或等于或小于250nM,或等于或小于150nM,或等于或小于50nM),和测定HLA匹配的新表位的总数(例如至少50,或至少100,或至少200,或至少300等)。
此外,考虑了这样的方法可进一步包括通过突变符号(mutation signature)(例如用于UV诱导的DNA损伤或吸烟诱导的DNA损伤的符号特征)过滤HLA匹配的新表位的步骤。在期望的情况下,还考虑了本文提出的方法可进一步包括使用组学数据来检测患病组织中的微卫星不稳定性(MSI)和/或错配修复缺陷(MMR)的步骤。
因此,并且从不同的角度来看,本发明人还考虑预测肿瘤对检查点抑制剂的阳性治疗反应的方法。这样的方法通常将包括从患者获得来自肿瘤组织和匹配的正常组织的组学数据,并且使用组学数据确定多个基于错义的患者特异性和肿瘤新表位的步骤。在进一步的步骤中,过滤并定量新表位以获得HLA匹配的新表位。在又一个步骤中,一旦确定HLA匹配的新表位的量已超过预定阈值量时,确定肿瘤对用检查点抑制剂治疗有反应。
类似地,本发明人还考虑了预测肿瘤对检查点抑制剂的阳性治疗反应的方法,其中从患者获得来自肿瘤组织和匹配的正常组织的组学数据,并且然后使用该组学数据确定多个基于错义的患者特异性和肿瘤特异性新表位。在另一步骤中,过滤新表位以获得HLA匹配的新表位,并且定量HLA匹配的新表位。在进一步的步骤中,测定定量的HLA匹配的新表位的突变符号,然后采用新表位的量和突变符号作为肿瘤对检查点抑制剂的阳性治疗反应的决定因素。
本发明主题的各种目的、特征、方面和优点将从以下对优选实施方案的详述以及附图中变得更加明显,附图中相同的附图标记表示相同的组分。。
附图说明
图1是各种癌症中新表位频率和编码变体频率的示例性图示以及各种癌症中独特新表位的频率的图示。
图2是新表位频率及其在癌症中表达成RNA的示例性图示。
图3是各种癌症的HLA匹配的新表位的频率的示例性图示。
图4是单一癌症类型(TNBC)的新表位和联合癌症的HLA限制性新表位的各种过滤过程的效果的示例性图示。还示例性显示了新表位相对于癌症驱动基因和非癌症基因的位置。
具体实施方式
本发明人现在已经发现HLA匹配的患者特异性和癌症特异性新表位能够用作用于一个或多个检查点抑制剂可能治疗肿瘤的成功的替代性指标,其中这样的新表位的数量高于阈值水平。这种较大数量的表达新表位可能归因于各种原因或与各种原因相关,包括MMR和/或MSI。另外,本发明人还发现,当新表位与能够产生独特的和肿瘤特异性的抗原的特定的突变模式(例如UV诱导的DNA损伤或吸烟诱导的DNA损伤)相关时,患者特异性和癌症特异性新表位可能指示可能的治疗成功。
新表位能够表征为产生独特的肿瘤特异性抗原的肿瘤细胞中表达的随机突变。因此,从不同的角度来看,可以通过考虑突变的类型(例如缺失、插入、颠换、转换、易位)和影响(例如无义、错义、移码等)来鉴定新表位,其可以因此作为第一内容过滤器,通过其消除沉默和其他不相关(例如不表达)突变。应该进一步理解的是,新表位序列能够被定义为具有较短长度的序列延伸(stretch)(例如7-11聚体),其中这样的延伸将包括氨基酸序列中的改变。最典型地,改变的氨基酸将位于或靠近中心氨基酸位置。例如,典型的新表位可具有A4-N-A4、或A3-N-A5、或A2-N-A7、或A5-N-A3、或A7-N-A2的结构,其中A是蛋白氨基酸而N是改变的氨基酸(相对于野生型或相对于匹配的正常)。例如,本文所考虑的新表位序列包括具有较短长度的序列延伸(例如5-30聚体,更典型地为7-11聚体,或12-25聚体),其中这样的延伸包括氨基酸序列中的改变。
因此,应该理解,取决于改变的氨基酸的位置,可以在包括改变的氨基酸的许多新表位序列中呈递单个氨基酸改变。有利的是,这样的序列可变性允许新表位的许多选择,并且因此增加可能有用的靶标的数量,然后能够基于一种或多种期望的性状(例如最高的针对患者HLA型的亲和力,最高的结构稳定性等)选择可能有用的靶标。最典型的是,新表位将被计算为长度为2-50个氨基酸,更典型地5-30个氨基酸,最典型地9-15个氨基酸,其中改变的氨基酸优选位于中央或以其他方式位于改善其与MHC结合的方式。例如,在MHC-I复合体呈递表位的情况下,典型新表位的长度会为约8-11个氨基酸,而用于经有MHC-II复合体呈递的典型新表位的长度会是长度为约13-17个氨基酸。如将容易理解的,由于新表位中改变的氨基酸的位置可以是中央以外,所以实际的肽序列以及新表位的实际拓扑结构可能显著不同。
当然,应该认识到,新表位的鉴定或发现可以从多种生物材料开始,包括新鲜活检组织、冷冻或以其他方式保存的组织或细胞样品、循环肿瘤细胞、外排体、各种体液(特别是血液)等。因此,合适的组学分析方法包括核酸测序,特别是在DNA上操作的NGS方法(例如Illumina测序、离子激流(ion torrent)测序、454焦磷酸测序、纳米孔测序等)、RNA测序(例如RNAseq、基于逆转录的测序等)以及蛋白质测序或基于质谱的测序(例如SRM、MRM、CRM等)。
因此,特别是对于基于核酸的测序,应特别认识到肿瘤组织的高通量基因组测序将允许快速鉴定新表位。然而,必须认识到,如果将如此获得的序列信息与标准参考比较,则正常发生的患者间变异(例如,由于SNP、短插入缺失、不同重复数量等)以及杂合性将导致相当大量的潜在假阳性新表位。值得注意的是,当将患者的肿瘤样品与同一患者的匹配的正常(即非肿瘤)样品进行比较时,能够消除这种不准确性。
在本发明主题的一个特别优选的方面,通过肿瘤和匹配的正常样品两者的全基因组测序和/或外显子组测序(典型在至少10x、更典型至少20x的覆盖深度)进行DNA分析。或者,也可以从先前序列测定中的已经建立的序列记录(例如,SAM,BAM,FASTA,FASTQ或VCF文件)提供DNA数据。因此,数据集可以包括未处理或已处理的数据集,并且示例性数据集包括具有BAMBAM格式、SAMBAM格式、FASTQ格式或FASTA格式的数据集。然而,特别优选的是,以BAMBAM格式或者作为BAMBAM diff对象提供数据集(参见例如US2012/0059670A1和US2012/0066001A1)。此外,应该注意的是,数据集反映了同一患者的肿瘤和匹配的正常样品,以获得患者和肿瘤特定信息。因此,可以排除不引起肿瘤的基因生殖细胞系变化(例如沉默突变、SNP等)。当然,应该认识到肿瘤样品可以来自最初的肿瘤、来自治疗开始时的肿瘤、来自复发的肿瘤或转移部位等。在大多数情况下,患者的匹配的正常样品可以是血液或来自与肿瘤相同组织类型的未患病组织。
类似地,可以以许多方式进行序列数据的计算分析。但是,在最优选的方法中,例如,如US 2012/0059670A1和US 2012/0066001A1中所公开的,使用BAM文件和BAM服务器通过位置引导的肿瘤和正常样品的同步比对来在计算机上进行分析。这样的分析有利地减少了假阳性新表位并显著减少了对存储器和计算资源的需求。
应该注意的是,任何针对计算机的语言都应该被理解为包括计算设备的任何适当组合,包括服务器、接口、系统、数据库、代理、对等设备、引擎、控制器或其他类型的单独或集体操作的计算设备。应该理解,计算设备包括被配置为执行存储在有形的非暂时性计算机可读存储介质(例如硬盘驱动器、固态驱动器、RAM、闪存、ROM等)上的软件指令的处理器。软件指令优选地配置计算设备以提供如下面关于所公开的装置所讨论的角色、责任或其他功能。此外,所公开的技术可以体现为计算机程序产品,其包括存储软件指令的非暂时性计算机可读介质,所述软件指令使得处理器执行与基于计算机的算法、进程、方法或其他指令。在特别优选的实施方案中,各种服务器、系统、数据库或接口可能基于HTTP、HTTPS、AES、公用密钥交换、网络服务API、已知金融交易协议或其他电子信息交换方法使用标准化协议或算法来交换数据。设备之间的数据交换可以通过分组交换网络、互联网、LAN、WAN、VPN或其他类型的分组交换网络;电路交换网络;小区交换网络;或其他类型的网络进行。
从不同的角度来看,可以建立具有预定长度的5至25个氨基酸并且包括至少一个改变的氨基酸的患者特异性和癌症特异性的计算机序列集合。这样的集合通常针对每个改变的氨基酸包括至少两个、至少三个、至少四个、至少五个或至少六个成员,其中改变的氨基酸的位置不相同。然后,这样的集合可以用于如下文中更详细描述的进一步过滤(例如通过亚细胞定位、转录/表达水平、MHC-I和/或II亲和力等)。
例如,并且使用对肿瘤和匹配的正常序列数据的同步定位指导分析,本发明人先前鉴定了来自各种癌症和患者的各种癌症新表位,包括以下癌症类型:BLCA,BRCA,CESC,COAD,DLBC,GBM,HNSC,KICH,KIRC,KIRP,LAML,LGG,LIHC,LUAD,LUSC,OV,PRAD,READ,SARC,SKCM,STAD,THCA,和UCEC。所有新表位数据可见于国际申请PCT/US16/29244中,其通过引用并入本文。
根据癌症的类型和阶段,应该注意的是,当检查点抑制剂给予患者时,并非所有确定的新表位都必然导致患者在治疗上等效的反应。事实上,本领域众所周知的是,只有一部分新表位会产生免疫应答。为了增加治疗上理想的应答的可能性,可以进一步过滤新表位。当然,应该理解的是下游分析不需要考虑用于本文呈现的方法的目的的沉默突变。然而,除了突变类型(例如缺失、插入、颠换、转换、易位)之外,优选的突变分析还提供突变影响(例如无义、错义等)的信息,并且因此可以作为通过其消除沉默突变的第一个内容过滤器。例如,当突变是移码、无义和/或错义突变时,可以选择新表位用于进一步的考虑。
在进一步的过滤方法中,还可以使新表位进行针对亚细胞定位参数的详细分析。例如,如果新表位被鉴定为具有膜相关位置(例如,位于细胞的细胞膜的外部)和/或如果计算机结构计算证实了新表位可能是溶剂暴露的,或呈现结构稳定的表位(例如J Exp Med 2014)等可以选择新表位序列用于进一步的考虑。
至于过滤新表位,一般考虑的是在组学(或其他)分析显示新表位实际表达的情况下新表位特别适用于本文。新表位的表达和表达水平的鉴定可以以本领域已知的所有方式进行,并且优选的方法包括定量RNA(hnRNA或mRNA)分析和/或定量蛋白质组学分析。最典型地,包含新表位的阈值水平将是相应匹配的正常序列表达水平的至少20%、至少30%、至少40%或至少50%的表达水平,从而确保(新)表位至少对免疫系统可能是“可见的”。因此,一般优选组学分析还包括基因表达分析(转录组分析),以便帮助鉴定具有突变的基因的表达水平。
本领域已知有许多转录组学分析方法,并且所有已知方法均被认为适用于本文。例如,优选的材料包括mRNA和初级转录物(hnRNA),并且RNA序列信息可以从反转录的多A+-RNA获得,所述反转录多聚A+-RNA又从相同患者的肿瘤样品和匹配的正常(健康)样品获得。同样,应当注意的是,虽然多A+-RNA通常优选作为转录组的代表,其它RNA形式(hn-RNA、非多腺苷酸化RNA、siRNA、miRNA等)也被认为适用于本文。优选的方法包括定量RNA(hnRNA或mRNA)分析和/或定量蛋白质组学分析,特别包括RNAseq。在其它方面,尽管各种替代方法(例如基于固相杂交的方法)也被认为是合适的,但使用基于RNA-seq、qPCR和/或rtPCR的方法进行RNA定量和测序。从另一角度来看,转录组学分析可能适合(单独或与基因组分析相结合)鉴定和定量具有癌症特异性和患者特异性突变的基因。
类似地,可以以许多方式进行蛋白质组学分析以确定新表位的RNA的实际翻译,并且本文考虑了所有已知的蛋白质组学分析方式。然而,特别优选的蛋白质组学方法包括基于抗体的方法和质谱方法。此外,应当注意的是,蛋白质组学分析不仅可以提供有关蛋白质本身的定性或定量信息,还可以包括蛋白质具有催化或其他功能活性的蛋白质活性数据。用于进行蛋白质组测定的一种示例性技术描述于US 7473532中,其通过引用并入本文。鉴定和甚至定量蛋白质表达的其它合适方法包括各种质谱分析(例如选择性反应监测(selective reaction monitoring,SRM)、多反应监测(multiplereaction monitoring,MRM)和连续反应监测(consecutive reaction monitoring,CRM))。
在过滤的又一个方面,可以将新表位针对含有已知人序列(例如患者或患者集合的序列)的数据库进行比较,以避免使用人类相同序列。此外,过滤还可以包括除去在SNP存在于肿瘤和匹配的正常序列中的患者中由SNP引起的新表位序列。例如,dbSNP(单核苷酸多态性数据库)是美国国家生物技术信息中心(NCBI)与美国国家人类基因组研究所(NHGRI)合作开发和主办的不同物种内和跨不同物种的遗传变异的免费公共档案库。尽管数据库的名称仅包含一类多态性(单核苷酸多态性(SNP))的集合,但它实际上包含了相对广泛的分子变异:(1)SNP,(2)短缺失和插入多态性(插入缺失/DIP),(3)微卫星标志物或短串联重复(STR),(4)多核苷酸多态性(MNP),(5)杂合序列,和(6)命名变体。dbSNP接受明显的中性多态性、对应于已知表型的多态性和没有变异的区域。
使用如上所述的这样的数据库和其他过滤选项,可以过滤患者和肿瘤特异性新表位以去除那些已知序列,产生具有显著减少的假阳性的多个新表位序列的序列集。
尽管如此,尽管过滤,但应该认识到,并非所有新表位都能被免疫系统看到,因为新表位也需要呈递在患者的MHC复合物上。事实上,只有一小部分新表位具有足够的表达亲和力,并且MHC复合物的大量多样性将排除使用大多数(如果不是全部的话)常见的新表位。因此,在免疫治疗的情况下,因此应该很明显的是,在新表位与MHC复合物结合并呈递时新表位更可能有效。从另一角度来看,用检查点抑制剂治疗成功需要多个新表位通过MHC复合物呈递,其中新表位必须对患者的HLA型具有最小亲和力。因此,应该理解的是,有效的结合和呈递是患者的新表位的序列和特定HLA类型的组合功能。最典型地,HLA型确定包括至少三种MHC-I亚型(例如HLA-A、HLA-B、HLA-C)和至少三种MHC-II亚型(例如HLA-DP、HLA-DQ、HLA-DR),优选其中每种亚型被确定为至少4位深度。然而,本文还考虑了更大的深度(例如6位、8位)。
一旦确定了患者的HLA类型(使用已知化学或计算机确定),就计算或从数据库获得HLA型结构解决方案,然后将其用于计算机对接模型以确定(通常过滤的)新表位对HLA结构解决方案的结合亲和力。如下面将进一步讨论的,用于确定结合亲和力的合适系统包括NetMHC平台(参见例如Nucleic Acids Res.2008Jul 1;36(Web Server issue):W509–W512.)。然后选择针对预先确定的HLA型的具有高亲和力(例如小于100nM、小于75nM、小于50nM)的新表位与MHC-I/II亚型的知识一起用于建立疗法。
可以使用本领域众所周知的湿化学中的各种方法进行HLA确定,并且所有这些方法都被认为适用于本文。然而,在特别优选的方法中,还可以使用如下文更详细所示的含有大部分或全部已知和/或常见HLA型的参考序列,通过计算机从组学数据预测HLA型。
例如,在根据本发明主题的一个优选方法中,通过数据库或测序机器提供作图至染色体6p21.3(或在其附近/在其发现HLA等位基因的任何其他位置)的相对大量的患者序列读段。最典型的序列读段将具有大约100-300个碱基的长度并包含元数据,包括读段质量、比对信息、方向、位置等。例如,合适的格式包括SAM、BAM、FASTA、GAR等。虽然不限于本发明主题,但通常优选患者序列读段提供至少5倍、更通常至少10倍、甚至更通常至少20倍、最通常至少30倍的覆盖深度。
除了患者序列读段之外,考虑的方法还使用一个或多个参考序列,其包括多个已知和不同HLA等位基因的序列。例如,典型的参考序列可以是合成的(没有相应的人类或其他哺乳动物对应物)序列,其包括至少一种具有该HLA型的多个HLA-等位基因的HLA-型的序列区段。例如,合适的参考序列包括HLA-A的至少50个不同等位基因的已知基因组序列的集合。备选地或另外地,参考序列还可以包括HLA-A的至少50个不同等位基因的已知RNA序列的集合。当然,并且如下面更详细地进一步讨论的,参考序列不限于HLA-A的50个等位基因,而是可以具有关于HLA型和等位基因的数量/组成的替代组成。最典型地,参考序列将以计算机可读格式存在,并将从数据库或其他数据存储设备提供。例如,合适的参考序列格式包括FASTA、FASTQ、EMBL、GCG或GenBank格式,并且可以从公共数据库(例如,IMGT、国际ImMunoGeneTics信息系统或等位基因频率网络数据库,EUROSTAM、URL:www.allelefrequencies.net)直接获得或构建。或者,也可以基于一个或多个预定标准,例如等位基因频率、种族等位基因分布、常见或罕见等位基因类型等,从个体已知HLA等位基因构建参考序列。
使用参考序列,患者序列读段现在可以通过de Bruijn图表进行拼接(thread)来鉴定最佳拟合的等位基因。在这种情况下,应当注意的是,每个个体携带每种HLA型的两个等位基因,并且这些等位基因可能非常相似,或者在某些情况下甚至是相同的。这种高度相似性对于传统的比对方案而言是一个重大问题。本发明人现在已经发现HLA等位基因以及甚至非常密切相关的等位基因可以使用构建de Bruijn图的方法来解析,其中通过将序列读段分解成相对较小的k聚体(通常具有10至20个碱基的长度)并且通过实施加权投票过程构建de Bruijn图,在所述投票过程中每个患者序列读段基于与该等位基因的序列匹配的序列读段的k聚体为每个等位基因提供投票(“定量读段支持”)。累积投票最高的等位基因表示最有可能预测的HLA等位基因。此外,通常优选的是,与等位基因匹配的每个片段也被用于计算该等位基因的总覆盖和覆盖深度。
可根据需要进一步提高或改进评分,尤其是在许多最高命中是相似的情况下(例如,在它们的评分的重要部分来自高度共享的k聚体集合的情况下)。例如,评分改进可以包括加权方案,其中将与当前最高命中基本相似(例如>99%,或其他预定值)的等位基因从未来的考虑中移除。然后通过一个因子(例如0.5)对由当前最高命中使用的k聚体的计数进行重新加权,并且通过将这些加权计数相加来重新计算每个HLA等位基因的评分。重复这个选择过程以找到新的最高命中。使用允许鉴定肿瘤表达的等位基因的RNA序列数据可以进一步提高该方法的准确度,所述等位基因有时可能仅仅是存在于DNA中的2个等位基因中的1个。在所考虑的系统和方法的进一步的有利方面,DNA或RNA或DNA和RNA二者的组合可以被处理以进行高度准确的HLA预测并且可以衍生自肿瘤或血液DNA或RNA。在进一步的方面,用于高准确度计算机HLA分型的合适方法和考虑因素描述于国际PCT/US16/48768中,其通过引用并入本文。
一旦鉴定出患者和肿瘤特异性新表位和HLA型,可以通过将新表位与HLA对接并确定最佳结合物(例如最低KD,例如小于500nM、或小于250nM、或小于150nM或小于50nM),例如使用NetMHC,进行进一步的计算分析。应该理解的是,这种方法不仅将鉴定对于患者和肿瘤是真实的特定新表位,而且还将鉴定最有可能呈递在细胞上并且因此最有可能引发具有治疗效果的免疫应答的那些新表位。当然,还应该理解的是,如下面进一步讨论的,如此鉴定的HLA匹配的新表位可以在将编码该表位的核酸作为有效载荷包含在病毒中之前在体外进行生化鉴定。
当然,应该理解的是,可以使用除NetMHC以外的系统来完成患者的HLA类型与患者特异性和癌症特异性新表位的匹配,并且合适的系统包括NetMHC II、NetMHCpan、IEDB分析资源(URL immuneepitope.org)、RankPep、PREDEP、SVMHC、Epipredict、HLABinding等(参见例如J Immunol Methods 2011;374:1–4)。在计算最高亲和力时,应该注意可以使用其中改变的氨基酸的位置发生移动的新表位的序列集合(同上)。备选地或另外地,对新表位的修饰可通过添加N-和/或C-末端修饰来进一步增加所表达的新表位与患者的HLA-型的结合。因此,新表位可以是经鉴定的天然蛋白或进一步修饰以更好匹配特定HLA型。而且,如果需要,可以计算相应的野生型序列(即没有氨基酸变化的新表位序列)的结合,以确保高差别亲和力。例如,新表位与其相应的野生型序列之间的MHC结合的特别优选的高差别亲和力为至少2倍、至少5倍、至少10倍、至少100倍、至少500倍、至少1000倍等)。
基于进一步的观察结果(数据未显示),本发明人考虑也可分析患者的组学数据(优选使用外显子组的同步定位引导比对或全基因组测序)以鉴定特定突变的类型或模式,并且这种模式(特别是与如上所述的最小数量的新表位组合)可以进一步指示检查点抑制剂可能的治疗成功。例如,在新表位与典型的紫外线损伤(例如,串联CC>TT/GG>AA突变)的突变模式相关的情况下并且在存在其中超过50或70或100个HLA匹配的患者特异性和癌症特异性新表位的情况下,可能比没有突变模式的情况和/或在存在较少的HLA匹配的患者特异性和癌症特异性新表位的情况下更可能成功使用检查点抑制剂治疗。类似地,在新表位与典型的吸烟诱导的DNA损伤(例如,高频G>T突变)的突变模式相关的情况下并且在存在其中超过50或70或100个新表位的情况下,可能比没有突变模式的情况和/或在存在较少的HLA匹配的患者特异性和癌症特异性新表位的情况下更可能成功使用检查点抑制剂治疗。
此外,考虑了新表位的增加数量可能是由于各种潜在的病症或现象造成的。例如,假定细胞中MMR(DNA错配修复)系统的缺陷或异常功能可能引发更多数量的新表位,这可能导致多链断裂和更高的突变率和可能具有更高的计数的HLA匹配的患者特异性和癌症特异性新表位,这也可能被视为MSI(微卫星不稳定性)。因此,还考虑了来自全基因组和/或外显子组测序的MMR和/或MSI的观察结果可以用作增加数量的HLA匹配的患者特异性和癌症特异性新表位的替代性指标。优选使用已知的组学分析算法和数据可视化(例如使用圆形图)针对匹配的正常鉴定MMR和/或MSI。
一旦已经使用一种或多种如上所述的方法适当地过滤新表位,则可以对过滤后的新表位进行定量/计数。正如容易理解的那样,用免疫检查点抑制剂对癌症进行治疗有效的治疗取决于表达和呈递的新表位的存在。虽然不希望受任何特定理论或假设的束缚,但是本发明人通常考虑到仅表达和呈递的新表位的一部分将导致治疗反应,并且肿瘤可能具有异质的癌细胞群体,每个群体可能具有各自的新表位。因此,本发明人考虑用免疫检查点抑制剂治疗癌症将需要最小/阈值量的HLA匹配的患者特异性和癌症特异性新表位。基于对各种癌症数据的回顾性分析并且如下更详细地进一步讨论的,本发明人因此考虑到当HLA匹配的患者特异性和癌症特异性新表位的量已经超过预定阈值量时应将检查点抑制剂施用于患者。最典型地,预定阈值量为至少50个HLA匹配的患者特异性和癌症特异性新表位,或至少100个HLA匹配的患者特异性和癌症特异性新表位,至少150个HLA匹配的患者特异性和癌症特异性新表位。
从不同的角度看,应该理解的是,诊断癌症中相对高数量的突变本身并不能预测用检查点抑制剂治疗癌症时的治疗反应,因为大量的这种突变(a)可能例如由于沉默突变而不导致新表位,(b)可能具有相应匹配的正常序列,并且因此根本不存在新表位,(c)可能不表达,并且因此免疫系统不可见,(d)可能不与患者特异性MHC复合物结合,并且因此免疫系统不可见。相反,考虑的系统和方法高度可信地鉴定HLA匹配的患者特异性和癌症特异性新表位。相反,应该认识到的是,具有相对较低突变频率的癌症确实可以在其中患者突变翻译相对较高数目(例如至少50或100或150或200个等)的HLA匹配的患者特异性和癌症特异性新表位的患者中治疗。
关于合适的检查点抑制剂,考虑了干扰检查点信号传导的所有化合物和组合物(例如CTLA-4(CD152)或PD-1(CD279))被认为适用于本文。例如,特别优选的检查点抑制剂包括派姆单抗、纳武单抗和易普利姆玛。最典型地,检查点抑制剂将按照常规方案并如处方信息中所述进行施用。但是,应当注意的是,在检查点抑制剂是肽或蛋白质的情况下,这些肽和/或蛋白质也可以在患者中从任何合适的表达系统(单独或与新表位和/或共刺激分子组合)表达。此外,如本文所用,关于检查点抑制剂的术语“施用”是指给患者直接施用(例如由医生或其他批准的医学专业人员)或间接施用(例如引起或建议施用)检查点抑制剂。
在又一个考虑的方面,联合疗法可能适用于将检查点抑制剂与一种或多种抗癌治疗剂一起使用。在其他药剂中,特别优选的是可以用用核酸构建体遗传修饰的病毒治疗肿瘤,所述核酸构建体导致至少一种鉴定的新表位表达以增强对肿瘤的免疫应答。例如,合适的病毒包括腺病毒、腺相关病毒、甲病毒、疱疹病毒、慢病毒等。然而,腺病毒是特别优选的。而且,进一步优选病毒是复制缺陷型和非免疫原性病毒,其典型地通过靶向缺失选择的病毒蛋白质(例如E1、E3蛋白质)来完成。通过缺失E2b基因功能可以进一步增强这种所需的性质,并且如最近报道的使用基因修饰的人293细胞可以实现高滴度的重组病毒(例如J Virol.1998Feb;72(2):926–933)。最典型地,期望的核酸序列(用于从病毒感染的细胞中表达)处于本领域众所周知的合适调控元件的控制之下。或者,免疫疗法不需要依赖病毒,而是可以用核酸疫苗接种或导致新表位表达的其他重组载体(例如作为单肽、串联微基因等)来实现。
同样,除病毒表达载体以外的其他免疫治疗剂也被认为是合适的,并且包括表达嵌合抗原受体或高亲和力CD16受体的基因工程细胞(特别是各种免疫活性细胞)。例如,考虑的免疫治疗剂包括NK细胞(例如,可从NantKwest,9920Jefferson Blvd.Culver City,CA 90232商购获得的NK细胞、haNK细胞或taNK细胞)或基因修饰的T细胞(例如表达T-细胞受体)或用HLA匹配的患者特异性和癌症特异性新表位离体刺激的T细胞。或者,HLA匹配的患者特异性和癌症特异性新表位还可以作为肽、任选与载体蛋白结合的肽进行施用。
实施例
数据集:如下所示的各种癌症的TCGA WGS和RNAseq数据下载自University of California,Santa Cruz(UCSC)Cancer Genomics Hub(https://cghub.ucsc.edu/)。基于完整WGS数据的可用性来选择TCGA样品以帮助进行计算机HLA分型。可用时使用相应样品的RNAseq数据。
肿瘤变体和新表位的鉴定:以基本上如US 2012/0059670A1和US 2012/0066001A1中公开的方式,通过使用BAM文件对肿瘤和正常样品的定位指导同步比对来鉴定单核苷酸变体(SNV)和插入/缺失(插入缺失)。由于HLA-A等位基因主要结合9聚体肽片段,本发明人致力于鉴定9聚体新表位。通过产生衍生自鉴定的SNV或插入缺失的9聚体氨基酸串(即,每个9聚体在独特位置具有改变的氨基酸)的所有可能排列来鉴定新表位。作为减少特定新表位的可能的脱靶效应的手段,本发明人针对从每个已知的人类基因产生的所有可能的9聚体肽序列过滤了所有鉴定的新表位。另外,本发明人还过滤了来自dbSNP(URL:www.ncbi.nlm.nih.gov/SNP/)的单核苷酸多态性,以得到(account for)在测序数据内可能遗漏的稀有蛋白质序列。通过RNA表达以及观察到的编码变体的等位基因频率对新表位进行进一步排序,以抵消由肿瘤异质性引起的问题。
HLA分型:TCGA样品的HLA分型数据不可用;因此,本发明人基本上如PCT/US16/48768中所述使用WGS、RNAseq数据和HLA森林算法进行了计算机HLA分型。简言之,使用Burrows-Wheeler比对算法将测序读段与IMGT/HLA数据库(URL:www.ebi.ac.uk/ipd/imgt/hla/)内的每个不同HLA等位基因进行比对。根据碱基的保守性并将读段质量评分考虑在内给每个比对评分。然后,每个HLA等位基因将具有总评分,以说明每个读段对某个HLA等位基因的比对程度,并且将具有最高评分的等位基因选作初级等位基因分型。然后通过去除与初级等位基因分型完全匹配的读段来进行次级等位基因分型,然后对读段重新评分而不与初级等位基因比对。使用该过程,发明人获得了所有样品的HLA-A、HLA-B、HLA-C和HLA-DRB1的分型结果至至少4位的水平。
新表位-HLA亲和力测定:使用NetMHC 3.4(URL:www.cbs.dtu.dk/services/NetMHC-3.4/)预测新表位是否会结合特定HLA等位基因。为了减少复杂性空间,本发明人选择结合分析局限于HLA-A等位基因,因为它们是最充分表征的HLA等位基因并且具有最好的结合亲和力模型。由于NetMHC 3.4工具没有针对每个鉴定的HLA-A等位基因的模型,如果患者的HLA-A分型不适用于NetMHC 3.4,则选择HLA超型进行结合预测。具有预测的结合亲和力<500nM蛋白质浓度的新表位用于进一步分析。但是,其他更严格的结合标准(<250nM,或<150nM,或<50nM)也被认为是适当的。
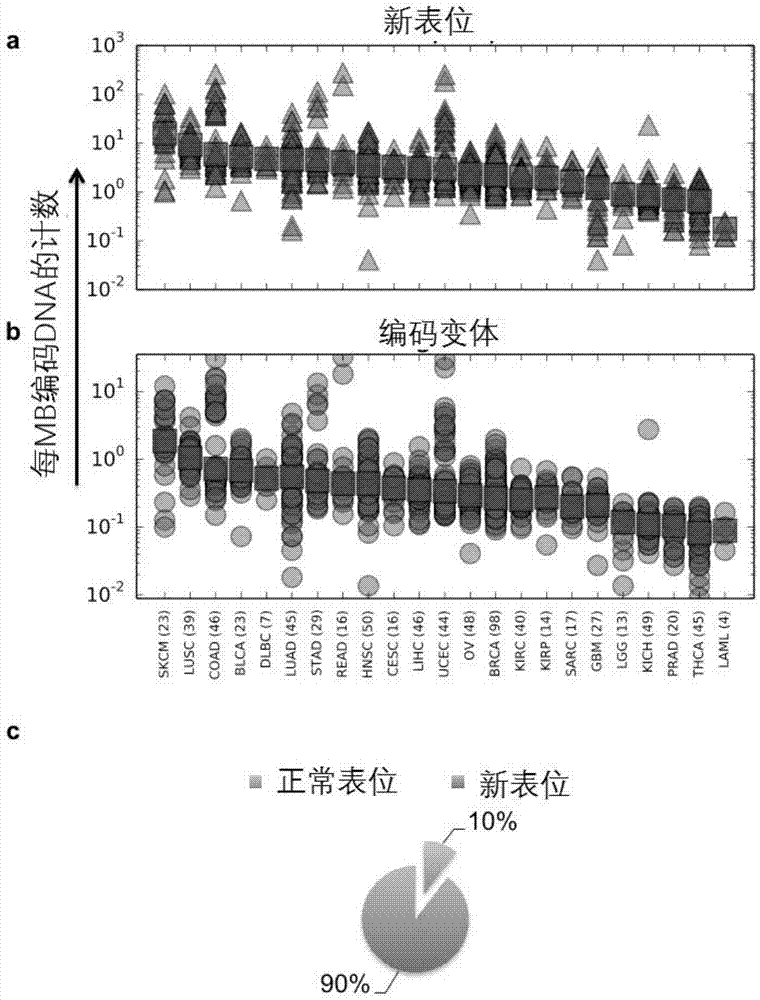
跨癌症类型的编码突变和新表位负荷:WGS数据和相应的RNAseq数据(如果可用)用于建立每兆碱基编码DNA的潜在新表位和体细胞编码变体的基线,所述编码DNA针对跨越23种癌症分类的750个患者样品,如图1所示。在此,TCGA内的跨23个癌症分类的750个患者样品显示新表位和变体计数。图(a)显示新表位计数;图(b)显示了变体计数。y轴显示每兆碱基编码DNA的计数(88MB用于人类基因组组装体(hg)19)。x轴显示每种癌症分类,其中患者数显示于括号中。中位样品计数由正方形表示。图(c)表示所有癌症类型内新表位和正常表位的百分比。
从图1可以很容易地看出,不同癌症类型的突变和新表位负荷不同,黑素瘤和鳞状细胞肺癌具有最高的新表位负荷,而甲状腺癌和急性骨髓性白血病具有最低的新表位负荷。针对已知人类序列的数据库过滤推定的新表位以消除潜在的脱靶效应显示,仅有10%的鉴定的新表位与已知蛋白质的片段相对应;因此,大多数突变产生独特的蛋白质序列。然而,即使独特的新表位的比例较高,表达和呈递也不能推定发生。事实上,正如下面更详细地显示的那样,应该认识到的是,表达和呈递的新表位的数量显著低于仅通过测序鉴定的新表位的数量。
新表位突变负荷和表达:由于I类MHC(MHC-I)呈递的表位长度,单一突变仍可导致许多不同的新表位表达。因此,含有数百个突变的个体患者肿瘤可能会含有数千个新表位。虽然许多肿瘤突变可能是乘客突变并且不负责癌症进展,但它们可能被用作治疗干预的靶标。RNAseq数据用于选择在所有癌症分类组合内,以及在具有不同突变负荷的癌症分类:子宫体子宫内膜癌、甲状腺癌和乳腺浸润癌内表达的新表位。最值得注意的是,由WGS鉴定的新表位计数与由多种癌症中的RNAseq鉴定的新表位表达相关(对于所有组合的癌症,皮尔森r=0.99),如图2所示。在此,图(a)描绘了所有癌症,图(b)描绘了甲状腺癌(THCA),图(c)描绘了乳腺浸润癌(BRCA),图(d)描绘了子宫体子宫内膜癌(UCEC)。y轴显示由WGS鉴定的每个样品的新表位的原始计数,并且x轴显示在由RNAseq确定的针对表达基因过滤后的新表位的原始计数。皮尔森相关系数、P值和样品编号显示在每个图上。无论平均突变负荷如何,具有高新表位负荷的癌症通常具有高新表位表达。
三阴性乳腺癌中新表位的鉴定:三阴性乳腺癌(TNBC)是一种侵袭性癌症,其治疗选择有限,标准化疗后进展后通常预后很差。TCGA数据集含有26个TNBC样品的WGS数据和RNAseq数据。TNBC中的新表位计数使用如下的迭代方法进行鉴定:根据WGS鉴定的编码变体预测每种可能的新表位;通过选择由RNAseq鉴定的表达的新表位来缩小新表位的数量;并且通过选择预测结合患者HLA型内的特定等位基因的新表位来进一步完善列表。新表位的这种选择性修剪产生了高质量的新表位,其对于每个患者是独特的,如表1所示。
表1
如图3(上图a)所示,对于所有26位患者,预测的新表位、表达的新表位和具有针对每种患者特异性HLA-A型的亲和力的新表位的数量分别为17,925、8184和228。显然,依赖测序确定的大量新表位(甚至对肿瘤相对于匹配正常的测序和分析)不能提供对检查点抑制剂治疗反应的有意义的预测因子。同样,对表达的进一步考虑将仅适度地去除假阳性结果,而将结果标准化为实际的HLA-结合物将显著增加表达和呈递的患者特异性和肿瘤特异性新表位的比例。
跨癌症分类的新表位的鉴定:由于在TNBC患者中没有共有的新表位,本发明人试图确定是否在TCGA数据集内的其他癌症分类之间共有任何新表位。为了确保常见的新表位也会与HLA复合物结合,本发明人将分析限于包含HLA-A*02:01等位基因的样品,所述样品在北美洲以高频发生。使用针对TNBC进行的相同迭代方法,本发明人鉴定了12个具有完整WGS和RNAseq数据的癌症的新表位,并且结果显示在图3中(下图b)。每个条内的阴影表示不同的患者样本。
在此,预测的新表位、表达的新表位和具有针对HLA-A*02:01的亲和力的新表位的数量分别为211,285、89,351和1,732。针对不同的样品大小校正,预测的新表位、表达的新表位和具有针对HLA-A*02:01的亲和力的新表位的平均数量分别为23,272、9,619和138。在这些数据中,鉴定出一种新表位发生在代表四种不同癌症类型的四种不同患者样品中:膀胱癌、尿路上皮癌、肺鳞状细胞癌、肺腺癌和乳腺浸润癌。代表两种不同癌症类型的患者对之间共有许多新表位(表2)。
表2
值得注意的是,仅基于WGS的初步新表位预测鉴定了TNBC患者中复发的几种新表位(数据未显示)。新表位-HLA结合分析后,由于HLA等位基因和患者结合潜能的差异,所有复发的新表位被消除。即使在组合的12种癌症分类中,复发性新表位也很罕见,只有4种不同癌症类型的患者共同检测到新表位,这再一次突出了对全面分子分型的需求。图3(下图饼状图c)进一步说明,在所有癌症中,约6%的新表位发生在癌症驱动基因中,这与先前的观察结果一致。
某些肿瘤如黑素瘤和肺癌具有高突变负荷,体细胞新表位的表达增加,应引起抗肿瘤反应并使这些癌症对检验点抑制剂更敏感。另一方面,突变/新表位相对较低的癌症应该不太可能具有表达/结合的新表位,因此应该对检查点抑制剂治疗的反应较差。不幸的是,这种假设过于简单化,并且对检查点抑制剂治疗的实际响应在很大程度上会取决于肿瘤特异性表达的新表位与患者的新表位的患者特异性亲和力与患者的HLA类型之间的患者和肿瘤特异性匹配。例如,虽然TCGA内的黑素瘤和肺癌样品具有高平均突变负荷,但是一些个体样品具有低的突变负荷。因此,应当理解的是,仅按疾病类型的普通分类将是过度包容性的,并且因此这样的受试者患者接受不可能有效的治疗。本发明人还鉴定了多种具有高突变负担的癌症类型的个体肿瘤样品,潜在地使患者对用检查点抑制剂治疗敏感。综合本发明人的发现表明,需要对患者肿瘤进行详细的分子分析以确定检查点抑制剂在这些药物的批准适应征之外的潜在益处。作为指导原则,鉴于以上提供的数据和考虑,分析应该集中在HLA匹配的(即对患者HLA型具有高亲和力的新表位,通常低于250nM,或低于150nM)必须存在阈值数量以上(例如至少50,更通常至少100)的患者特异性和癌症特异性新表位。
图4示例性描绘了如上所述鉴定的各种癌症新表位的变体计数。显而易见的是,某些癌症具有相对高数量的新表位,而其他癌症仅具有中等数量的新表位。此外,应当注意的是,同一类型癌症内新表位发生的变异性不均匀。实际上,一些癌症具有相对低的新表位的平均数量,但高度可变性远高于100的预定阈值(例如,HNSC、LUAD;阈值以虚线显示)。值得注意的是,具有高于阈值的新表位计数的癌症显示出对用检查点抑制剂(例如UCEC,READ,BLCA,SKCM,LUSC,COAD,STAD)治疗有反应的显著更高的可能性。此外,还观察到这些癌症也通常与MMR和/或MSI有关。
这里对数值范围的引用仅仅是用作单独提及落入该范围内的每个单独值的简略表达方法。除非在此另有指示,否则每个单独的值都并入说明书中,就好像它在此单独列举一样。本文描述的所有方法可以以任何合适的顺序执行,除非在此另有指示或者与上下文以其他方式明显矛盾。针对本文中的某些实施例提供的任何和所有示例或示例性语言(例如“诸如”)的使用仅意在更好地阐明本发明,而不是对本发明另外要求保护的范围进行限制。说明书中的任何语言都不应被解释为表示对本发明的实践必不可少的任何非要求保护的要素。
对于本领域技术人员来说显而易见的是,在不脱离本文的发明构思的情况下,除了已经描述的那些以外,还可以有更多的修改。因此,本发明的主题不限于所附权利要求的范围。而且,在解释说明书和权利要求书时,所有术语应该以与上下文一致的最宽泛可能的方式来解释。具体而言,术语“包含”和“包括”应该被解释为以非排他性方式引用元件、组件或步骤,指示所提及的元件、组件或步骤可以存在、或被利用、或与未明确引用的其他元件、组件或步骤进行组合。在说明书权利要求涉及选自由A、B、C......和N组成的组中的至少一项的情况下,该文本应该被解释为只需要来自该组的一个元件,而不是A+N或B+N等。
权利要求书(按照条约第19条的修改)
1.一种使用免疫疗法改善癌症治疗的方法,包括:
从患者获得来自肿瘤组织和匹配的正常组织的组学数据,并且使用所述组学数据确定多个基于错义的患者特异性和肿瘤特异性新表位;
从患者特异性和肿瘤特异性新表位中鉴定表达的新表位;
过滤所述表达的新表位以获得HLA匹配的新表位并定量HLA匹配的新表位;和
一旦确定HLA匹配的新表位的量已超过预定阈值量,向患者施用检查点抑制剂。
2.根据权利要求1所述的方法,其中使用多个不同的个体新表位序列对新表位中的每一个进行过滤表达的新表位的步骤,其中改变的氨基酸在新表位序列内具有不同的位置。
3.根据权利要求2所述的方法,其中所述个体新表位序列长度为7至20个氨基酸。
4.根据权利要求1所述的方法,其中过滤步骤进一步包括通过至少一个事先已知的分子变异进行过滤的步骤,所述分子变异选自由单核苷酸多态性、短缺失和插入多态性、微卫星标志物、短串联重复、杂合序列、多核苷酸多态性和命名变体组成的组。
5.根据权利要求1所述的方法,其中过滤步骤包括测定表达的新表位与该患者的至少一种I型MHC亚型和至少一种II型MHC亚型的亲和力。
6.根据权利要求1所述的方法,其中过滤步骤进一步包括测定表达的新表位的表达水平。
7.根据权利要求1所述的方法,其中HLA匹配的新表位与该患者的至少一种I型MHC亚型或至少一种II型MHC亚型的亲和力等于或小于150nM。
8.根据权利要求1所述的方法,其中定量HLA匹配的新表位包括定量新表位的步骤与该患者的至少一种I型MHC亚型或至少一种II型MHC亚型的亲和力,并测定HLA匹配的新表位的总数。
9.根据权利要求1所述的方法,其进一步包括通过突变符号过滤HLA匹配的新表位的步骤。
10.根据权利要求9所述的方法,其中突变符号是针对UV诱导的DNA损伤或吸烟诱导的DNA损伤的符号特征。
11.根据权利要求1所述的方法,其中HLA匹配的新表位的预定阈值量为至少100个HLA匹配的新表位。
12.根据权利要求11所述的方法,其中至少100个HLA匹配的新表位与该患者的至少一种I型MHC亚型或至少一种II型MHC亚型的亲和力等于或小于150nM。
13.根据权利要求1所述的方法,其进一步包括使用组学数据检测患病组织中的微卫星不稳定性(MSI)的步骤。
14.根据权利要求1所述的方法,其进一步包括使用组学数据检测患病组织中的错配修复缺陷(MMR)的步骤。
15.根据权利要求1所述的方法,其中所述检查点抑制剂是CTLA-4抑制剂或PD-1抑制剂。
16.预测肿瘤对检查点抑制剂的阳性治疗反应的方法,包括:
从患者获得来自肿瘤组织和匹配的正常组织的组学数据,并且使用组学数据确定多个基于错义的患者特异性和肿瘤特异性新表位;
从患者特异性和肿瘤特异性新表位中鉴定表达的新表位;过滤表达的新表位以获得HLA匹配的新表位,并定量HLA匹配的新表位;和
确定HLA匹配的新表位的量已超过预定阈值量时,确定肿瘤对用检查点抑制剂治疗有反应。
17.根据权利要求16所述的方法,其进一步包括通过突变符号过滤HLA匹配的新表位的步骤。
18.根据权利要求16所述的方法,其进一步包括使用组学数据检测患病组织中微卫星不稳定(MSI)和错配修复缺陷(MMR)中的至少一种的步骤。
19.预测肿瘤对检查点抑制剂的阳性治疗反应的方法,包括
从患者获得来自肿瘤组织和匹配的正常组织的组学数据,并且使用所述组学数据确定多个基于错义的患者特异性和肿瘤特异性新表位;
从患者特异性和肿瘤特异性新表位中鉴定表达的新表位;
过滤表达的新表位以获得HLA匹配的新表位,并定量HLA匹配的新表位;
鉴定所定量的HLA匹配的新表位的突变符号;和
使用新表位的量和突变符号作为肿瘤对检查点抑制剂的阳性治疗反应的决定因素。
20.根据权利要求19所述的方法,其中突变符号是针对UV诱导的DNA损伤或吸烟诱导的DNA损伤的特征。
说明或声明(按照条约第19条的修改)
国际局认为独立权利要求16和19相对于对比文件1(New England Journal of Medicine,第371卷,第2189-2199页(2014))不具有创造性。申请人不认同,特别是鉴于所做修改。
关于权利要求16和19,必须认识到对比文件1教导鉴定在具有相同的癌症并且已经经历长期用伊匹单抗治疗的益处的患者中共有的标志物新表位。基于先验知识的共用的新表位的鉴定用于指示用伊匹单抗对黑素瘤的成功治疗。此外,对比文件1教导了单独患者中新表位的阈值量没有指示成功治疗。对比文件1第2192页的右栏(“[A]高负荷单独不足以赋予临床益处,原因在于存在对治疗方法不反应的具有高变异负担的肿瘤”);对比文件1第2194页的右栏(“甚至来自具有超过1000个变异的患者(患者NR9521和NR4631)的肿瘤没有反应”)。实际上,对比文件1教导取决于在患者中共有的标志物新表位和癌症来预测治疗。因此,对比文件1没有教导修改后的权利要求16和19的主题。
具体地,对比文件1完全没有意识到经由检查点抑制剂的成功治疗能够预测为由单一患者的肿瘤和匹配的正常组学数据的分析而不必参照表示成功的新表位的先验知识。实际上,对比文件1没有教导对患者和患者中的癌症特异性的新表位(通过患者的HLA型来表达和显示)的定量。对比文件1也没有意识到这样的新表位的量(单独或对于每个新表位的组合物不可知)能够预测由检查点抑制剂对癌症的成功治疗。相反,对比文件1依赖于对表示成功药物治疗的癌症治疗结果和标志物新表位的特定组合物的先验知识。
尽管上述的对比文件1的缺陷,权利要求16和19已被修改为要求鉴定表达的新表位、然后过滤表达的新表位以发现HLA匹配的新表位。显而易见地,对比文件1没有教导该限制,事实上,注意到其没有这样做。对比文件1的第2194页右栏(“我们不能排除……存在生物上相关的表位,其在生物信息学上被预测,但在患者中没有被表达或呈现”)。因此,对比文件1没有教导权利要求16和19的主题或者给予这样的动机。
鉴于本修改和陈述,申请人认为现在所有权利要求处于被授权的条件。因此在这种情况下申请人要求给予肯定性的IPRP。
- 还没有人留言评论。精彩留言会获得点赞!
- SPNP‑I在制备Nav1.7通道工具试剂中的应用的制造方法与工艺
- SPNP‑I在制备Nav1.2通道工具试剂中的应用的制造方法与工艺
- SPNP‑IX在制备Nav1.3通道抑制剂中的应用的制造方法与工艺
- SPNP‑IX在制备Kv4.2通道工具试剂中的应用的制造方法与工艺
- SPNP‑IX在制备Kv4.3通道工具试剂中的应用的制造方法与工艺
- SPNP‑27在制备Nav1.5通道抑制剂中的应用的制造方法与工艺
- SPNP‑27在制备Nav1.3通道抑制剂中的应用的制造方法与工艺
- 一种多肽在制备Nav1.3钠通道工具试剂中的应用的制造方法与工艺
- SPKP‑XI在制备Kv4.2通道抑制剂中的应用的制造方法与工艺
- SGLT‑2糖尿病抑制剂及其中间体的制备方法与流程