基于血小板差异表达基因标记的分类方法、装置及系统与流程
本发明涉及计算机技术领域,具体涉及一种基于血小板差异表达基因标记的分类方法、装置及系统。
背景技术:
转录组是细胞内所有基因产生的信使核糖核酸的集合,对细胞的正常运转有重要的作用。在人类或其他生物体内,几乎所有的细胞都含有同样的一套基因,但是,其表达的模式却与细胞的类型、所属的器官组织、所处的生理条件或所在的样本个体密切相关,因此,通过解读和比较转录组提供的信息,研究人员能够鉴别出两组具有不同属性的个体之间的差异表达基因标记,并基于差异表达基因标记的表达模式,对未知的细胞或组织或个体进行准确的分类。
血小板是血液的重要组成部分,其主要功效是凝聚于血管伤口处止血。血小板细胞来源于骨髓细胞,并不含有细胞核。长期以来,人们普遍理解为血小板内核糖核酸的种类和表达谱比较稳定,但最新的一系列研究表明血小板有可能在对于某些疾病包括肿瘤的全身性或局部性反应中起重要作用。通过对外界刺激信号的应答或者直接吞噬循环系统内的信使核糖核酸,血小板转录组能够呈现出特异的表达谱,因而解读血小板转录组的基因标记特征在某些疾病早期诊断方面有广泛的应用前景。
目前,现有的差异表达基因标记的鉴定方法灵敏度较差,导致基于差异表达基因标记的未知个体分类结果精度不够,造成后续的应用受限。
如何能够快速、准确地鉴别出针对特定组群的差异表达基因标记,提高该组群相应个体的分类精确度,是本领域技术人员亟需解决的问题。
技术实现要素:
针对现有技术中的缺陷,本发明提供基于血小板差异表达基因标记的分类方法、装置及系统,能够快速、准确地鉴别出针对特定组群的差异表达基因标记,提高该组群相应个体的分类精确度。
第一方面,本发明提供一种基于血小板差异表达基因标记的分类方法,该方法包括:
信息获取步骤:获取目标个体的测序读取序列;
分析对比步骤:将所述测序读取序列和预获取的人类基因组进行对比分别,获取对比分析结果;
信息显示步骤:显示所述对比分析结果。
本发明提供另一种基于血小板差异表达基因标记的分类方法,该方法包括:正负样本集构建步骤:根据二元性特征,将预获取的目标样本分成正样本集和负样本集,正样本集和负样本集的数量均满足预定数量要求;
基因测序读取序列获取步骤:对正样本集和负样本集进行血小板转录组测序,分别获取正样本基因测序读取序列和负样本基因测序读取序列;
预处理步骤:将正样本基因测序读取序列与预获得的测序衔接序列进行比对,删除正样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始正样本测序读取序列;
检验初始正样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始正样本测序读取序列,形成正样本测序读取序列集合;
将负样本基因测序读取序列与测序衔接序列进行比对,删除负样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始负样本测序读取序列;
检验初始负样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始负样本测序读取序列,形成负样本测序读取序列集合;
基因表达量估算步骤:根据后缀阵列搜索算法和序列拆分/搜索/延伸策略,将正样本测序读取序列集合和负样本测序读取序列集合分别与预获取的人类基因组进行比对,分别获取正样本测序读取序列对比结果和负样本测序读取序列对比结果;
根据期望最大化算法和正样本测序读取序列对比结果,确定正样本基因表达估计值;
根据期望最大化算法和负样本测序读取序列对比结果,确定负样本基因表达估计值;
差异表达基因标记确定步骤:采用线性统计模型和经验贝叶斯方法,将正样本基因表达估计值和负样本基因表达估计值进行比较,获取表达差异值,并将表达差异值低于第三阈值的基因,作为差异表达基因标记;
超平面表达式构建步骤:获取每个差异表达基因标记在正样本集中的正样本基因表达估计值,作为正样本标记基因表达估计值;
获取每个差异表达基因标记在负样本集中的负样本基因表达估计值,作为负样本标记基因表达估计值;
根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式:其中,为系数,b为偏移量,为超平面的变量;
量化分类步骤:根据超平面表达式和预获取实体的基因表达量估计值获取该实体的量化分类结果,并根据显示模式,进行显示。
进一步地,根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式,具体包括:构建超平面表达式:使得其中,为第i个样本标记基因表达估计值,若yi=1表示第i个样本标记基因表达估计值为正样本标记基因表达估计值,若yi=-1表示第i个样本标记基因表达估计值为负样本标记基因表达估计值,m为第i个样本标记基因表达估计值所在的平面到超平面的距离。
进一步地,第一阈值为10%,第二阈值为50%,第三阈值为0.001。
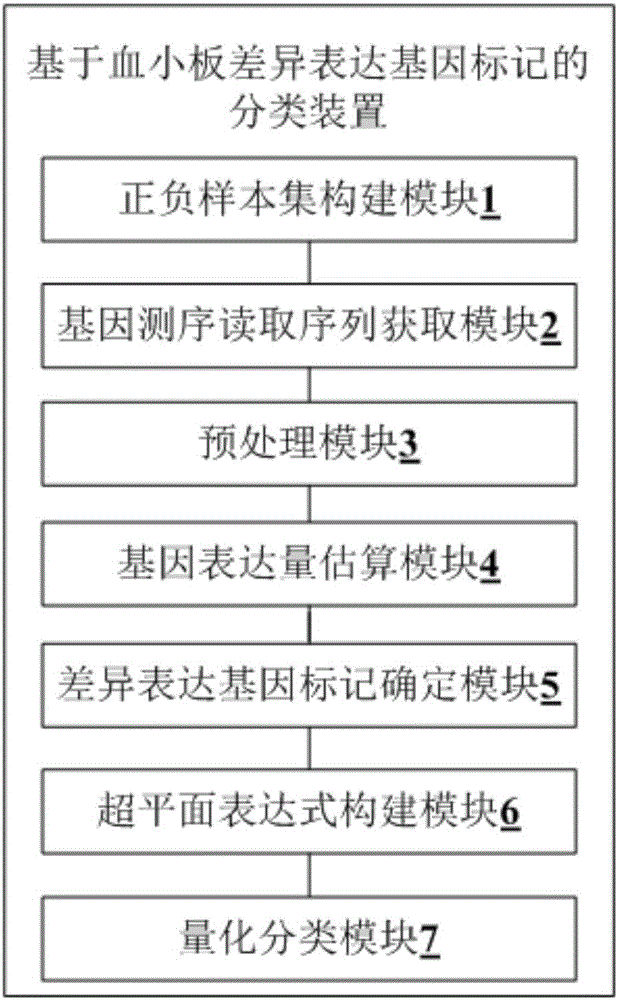
第二方面,本发明提供一种基于血小板差异表达基因标记的分类装置,该装置包括正负样本集构建模块、基因测序读取序列获取模块、预处理模块、基因表达量估算模块、差异表达基因标记确定模块、超平面表达式构建模块和量化分类模块,正负样本集构建模块用于根据二元性特征,将预获取的目标样本分成正样本集和负样本集,正样本集和负样本集的数量均满足预定数量要求;基因测序读取序列获取模块用于对正样本集和负样本集进行血小板转录组测序,分别获取正样本基因测序读取序列和负样本基因测序读取序列;预处理模块用于将正样本基因测序读取序列与预获得的测序衔接序列进行比对,删除正样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始正样本测序读取序列;检验初始正样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始正样本测序读取序列,形成正样本测序读取序列集合;将负样本基因测序读取序列与测序衔接序列进行比对,删除负样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始负样本测序读取序列;检验初始负样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始负样本测序读取序列,形成负样本测序读取序列集合;基因表达量估算模块用于根据后缀阵列搜索算法和序列拆分/搜索/延伸策略,将正样本测序读取序列集合和负样本测序读取序列集合分别与预获取的人类基因组进行比对,分别获取正样本测序读取序列对比结果和负样本测序读取序列对比结果;根据期望最大化算法和正样本测序读取序列对比结果,确定正样本基因表达估计值;根据期望最大化算法和负样本测序读取序列对比结果,确定负样本基因表达估计值;差异表达基因标记确定模块用于采用线性统计模型和经验贝叶斯装置,将正样本基因表达估计值和负样本基因表达估计值进行比较,获取表达差异值,并将表达差异值低于第三阈值的基因,作为差异表达基因标记;超平面表达式构建模块用于获取每个差异表达基因标记在正样本集中的正样本基因表达估计值,作为正样本标记基因表达估计值;获取每个差异表达基因标记在负样本集中的负样本基因表达估计值,作为负样本标记基因表达估计值;根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式:其中,为系数,b为偏移量,为超平面的变量;量化分类模块用于根据超平面表达式和预获取实体的基因表达量估计值获取该实体的量化分类结果,并根据显示模式,进行显示。
进一步地,超平面表达式构建模块在根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式时,具体用于:构建超平面表达式:使得其中,为第i个样本标记基因表达估计值,若yi=1表示第i个样本标记基因表达估计值为正样本标记基因表达估计值,若yi=-1表示第i个样本标记基因表达估计值为负样本标记基因表达估计值,m为第i个样本标记基因表达估计值所在的平面到超平面的距离。
进一步地,预处理模块中第一阈值为10%,预处理模块中第二阈值为50%,差异表达基因标记确定模块中第三阈值为0.001。
第三方面,本发明提供一种基于血小板差异表达基因标记的分类系统,该系统包括处理器和与处理器连接的测序平台、服务器和显示屏,显示屏上设有触摸屏,触摸屏与处理器连接,测序平台用于根据二元性特征,将预获取的目标样本分成正样本集和负样本集,对正样本集和负样本集进行血小板转录组测序,分别获取正样本基因测序读取序列和负样本基因测序读取序列,并传输至处理器,处理器用于获取正样本基因测序读取序列、负样本基因测序读取序列和实体的基因表达量估计值根据基于血小板差异表达基因标记的分类方法,获取超平面表达式和该实体的量化分类结果,并传送至服务器和显示屏,服务器用于存储超平面表达式和实体的量化分类结果,显示屏用于根据显示模式指令显示实体的量化分类结果,触摸屏用于接收用户点选的显示模式指令。
由上述技术方案可知,本实施例提供的基于血小板差异表达基因标记的分类方法、装置及系统,通过预处理步骤,能够有效去除噪音信息,有助于提高数据处理的精确度。该方法采用后缀阵列搜索算法和序列拆分/搜索/延伸策略,有助于提高序列对比的精确度。同时,该方法能够根据正样本基因表达估计值和负样本基因表达估计值获得超平面表达式,有助于快速、准确地处理不同实体的基因表达量估计值,并获取准确、直观的量化分类结果。
因此,本实施例基于血小板差异表达基因标记的分类方法、装置及系统,能够快速、准确地鉴别出特定群组的差异表达基因标记,且对该群组实体的分类判别准确、可靠,数据处理效率高。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍。在所有附图中,类似的元件或部分一般由类似的附图标记标识。附图中,各元件或部分并不一定按照实际的比例绘制。
图1示出了本发明所提供的一种基于血小板差异表达基因标记的分类方法流程图;
图2示出了本发明所提供的一种基于血小板差异表达基因标记的分类装置的结构框图;
图3示出了本发明所提供的一种基于血小板差异表达基因标记的分类系统的结构示意图。
具体实施方式
下面将结合附图对本发明技术方案的实施例进行详细的描述。以下实施例仅用于更加清楚地说明本发明的技术方案,因此只是作为示例,而不能以此来限制本发明的保护范围。
需要注意的是,除非另有说明,本申请使用的技术术语或者科学术语应当为本发明所属领域技术人员所理解的通常意义。
本发明实施例所提供的一种基于血小板差异表达基因标记的分类方法,该方法包括:
信息获取步骤:获取目标个体的测序读取序列;
分析对比步骤:将所述测序读取序列和预获取的人类基因组进行对比分别,获取对比分析结果;
信息显示步骤:显示所述对比分析结果。
本发明实施例提供另一种基于血小板差异表达基因标记的分类方法,结合图1,该方法包括:
正负样本集构建步骤S1:根据二元性特征,将预获取的目标样本分成正样本集和负样本集,正样本集和负样本集的数量均满足预定数量要求,在此,二元性特征是预先给定或关注的特征,并且,正样本集中的正样本数量和负样本集中的负样本数量需相同,或相对接近。
基因测序读取序列获取步骤S2:对正样本集和负样本集进行血小板转录组测序,分别获取正样本基因测序读取序列和负样本基因测序读取序列。
预处理步骤S3:将正样本基因测序读取序列与预获得的测序衔接序列进行比对,删除正样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始正样本测序读取序列,在此,该方法采用半全局对齐算法的cutadapt程序进行比对,该cutadapt程序的版本号为1.8.1。
检验初始正样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始正样本测序读取序列,形成正样本测序读取序列集合,在此,第一阈值为10%,第二阈值为50%,如一个100个碱基长度的初始正样本测序读取序列含有超过10个未知碱基,该序列在后续分析中将不被采用,由于初始正样本测序读取序列中的低质量碱基,如Q≤10的碱基,代表噪音信息的概率较大,弃用所有含有低质量碱基个数超过50%的初始正样本测序读取序列,如果一个100个碱基长度的初始正样本测序读取序列含有超过50个低质量碱基,该序列在后续分析中将不被采用。
将负样本基因测序读取序列与测序衔接序列进行比对,删除负样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始负样本测序读取序列。
检验初始负样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始负样本测序读取序列,形成负样本测序读取序列集合。
基因表达量估算步骤S4:根据后缀阵列搜索算法和序列拆分/搜索/延伸策略,将正样本测序读取序列集合和负样本测序读取序列集合分别与预获取的人类基因组进行比对,分别获取正样本测序读取序列对比结果和负样本测序读取序列对比结果。
根据期望最大化算法和正样本测序读取序列对比结果,确定正样本基因表达估计值。
根据期望最大化算法和负样本测序读取序列对比结果,确定负样本基因表达估计值。
差异表达基因标记确定步骤S5:采用线性统计模型和经验贝叶斯方法,将正样本基因表达估计值和负样本基因表达估计值进行比较,获取表达差异值,并将表达差异值低于第三阈值的基因,作为差异表达基因标记,在此,线性统计模型为limma程序包构建的模型,其中,第三阈值为0.001,在此,若某正样本基因的表达估计值集合中80%的数值小于第四阈值,例如第四阈值为5,则该正样本基因表达估计值的集合需要删除,以有利于保证数据处理的精确性,且降低噪音信息的干扰。
超平面表达式构建步骤S6:获取每个差异表达基因标记在正样本集中的正样本基因表达估计值,作为正样本标记基因表达估计值。
获取每个差异表达基因标记在负样本集中的负样本基因表达估计值,作为负样本标记基因表达估计值。
根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式:其中,为系数,b为偏移量,为超平面的变量。
量化分类步骤S7:根据超平面表达式和预获取实体的基因表达量估计值获取该实体的量化分类结果,并根据显示模式,进行显示。
由上述技术方案可知,本实施例提供的基于血小板差异表达基因标记的分类方法,通过预处理步骤,能够有效去除噪音信息,有助于提高数据处理的精确度。该方法采用后缀阵列搜索算法和序列拆分/搜索/延伸策略,有助于提高序列对比的精确度。同时,该方法能够根据正样本基因表达估计值和负样本基因表达估计值获得超平面表达式,有助于快速、准确地处理不同实体的基因表达量估计值,并获取准确、直观的量化分类结果。
因此,本实施例基于血小板差异表达基因标记的分类方法,能够快速、准确地鉴别出特定群组的差异表达基因标记,且对该群组实体的分类判别准确、可靠,数据处理效率高。
同时,该方法可以应用于医疗领域,诊治医生可以针对该方法的量化分类结果进行分析,为判断检测对象体内是否存在癌症提供有效的信息支持,且数据处理效率高、判断结果准确。同时,检测对象没有任何伤害。
具体地,在根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式时,本实施例基于血小板差异表达基因标记的分类方法的具体实现过程如下:
构建超平面表达式:使得其中,为第i个样本标记基因表达估计值,若yi=1表示第i个样本标记基因表达估计值为正样本标记基因表达估计值,若yi=-1表示第i个样本标记基因表达估计值为负样本标记基因表达估计值,m为第i个样本标记基因表达估计值所在的平面到超平面的距离。在此,用y的取值表征该样本标记基因表达估计值的分类状况。同时,该方法构建的超平面表达式会使得正样本集和负样本集之间的距离最大,以达到正样本集和负样本集最好的分类效果。此时,要保证所以要最小化:
通过拉格朗日求导后,即要最大化:
其中,n为目标样本总数,c为系数,ci满足可得并通过网格寻优法,获取最优参数。
第二方面,本发明实施例提供一种基于血小板差异表达基因标记的分类装置,结合图2,该装置包括正负样本集构建模块1、基因测序读取序列获取模块2、预处理模块3、基因表达量估算模块4、差异表达基因标记确定模块5、超平面表达式构建模块6和量化分类模块7。
正负样本集构建模块1用于根据二元性特征,将预获取的目标样本分成正样本集和负样本集,正样本集和负样本集的数量均满足预定数量要求。
基因测序读取序列获取模块2用于对正样本集和负样本集进行血小板转录组测序,分别获取正样本基因测序读取序列和负样本基因测序读取序列。
预处理模块3用于将正样本基因测序读取序列与预获得的测序衔接序列进行比对,删除正样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始正样本测序读取序列;检验初始正样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始正样本测序读取序列,形成正样本测序读取序列集合;将负样本基因测序读取序列与测序衔接序列进行比对,删除负样本基因测序读取序列中与测序衔接序列比对一致的序列部分,形成初始负样本测序读取序列;检验初始负样本测序读取序列中的未知碱基比例和低质量碱基比例,保留未知碱基比例小于第一阈值,且低质量碱基比例小于第二阈值的初始负样本测序读取序列,形成负样本测序读取序列集合。其中,第一阈值为10%,第二阈值为50%。
基因表达量估算模块4用于根据后缀阵列搜索算法和序列拆分/搜索/延伸策略,将正样本测序读取序列集合和负样本测序读取序列集合分别与预获取的人类基因组进行比对,分别获取正样本测序读取序列对比结果和负样本测序读取序列对比结果;根据期望最大化算法和正样本测序读取序列对比结果,确定正样本基因表达估计值;根据期望最大化算法和负样本测序读取序列对比结果,确定负样本基因表达估计值。
差异表达基因标记确定模块5用于采用线性统计模型和经验贝叶斯装置,将正样本基因表达估计值和负样本基因表达估计值进行比较,获取表达差异值,并将表达差异值低于第三阈值的基因,作为差异表达基因标记。其中,第三阈值为0.001。
超平面表达式构建模块6用于获取每个差异表达基因标记在正样本集中的正样本基因表达估计值,作为正样本标记基因表达估计值;获取每个差异表达基因标记在负样本集中的负样本基因表达估计值,作为负样本标记基因表达估计值;根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式:其中,为系数,b为偏移量,为超平面的变量。
量化分类模块7用于根据超平面表达式和预获取实体的基因表达量估计值获取该实体的量化分类结果,并根据显示模式,进行显示。
由上述技术方案可知,本实施例提供的基于血小板差异表达基因标记的分类装置,通过预处理步骤,能够有效去除噪音信息,有助于提高数据处理的精确度。该装置采用后缀阵列搜索算法和序列拆分/搜索/延伸策略,有助于提高序列对比的精确度。同时,该装置能够根据正样本基因表达估计值和负样本基因表达估计值获得超平面表达式,有助于快速、准确地处理不同实体的基因表达量估计值,并获取准确、直观的量化分类结果。
因此,本实施例基于血小板差异表达基因标记的分类装置,能够快速、准确地鉴别出特定群组的差异表达基因标记,且对该群组实体的分类判别准确、可靠,数据处理效率高。
具体地,超平面表达式构建模块6在根据正样本标记基因表达估计值和负样本标记基因表达估计值,构建超平面表达式时,具体用于:构建超平面表达式:使得其中,为第i个样本标记基因表达估计值,若yi=1表示第i个样本标记基因表达估计值为正样本标记基因表达估计值,若yi=-1表示第i个样本标记基因表达估计值为负样本标记基因表达估计值,m为第i个样本标记基因表达估计值所在的平面到超平面的距离。在此,超平面表达式构建模块6能够正样本集和负样本集的样本标记基因表达估计值,以获取超平面表达式,有利于后续对实体的量化分类处理,得到直观、准确的量化表达结果。
第三方面,本发明实施例提供一种基于血小板差异表达基因标记的分类系统,结合图3,该系统包括处理器31和与处理器31连接的测序平台32、服务器33和显示屏34,显示屏34上设有触摸屏,触摸屏与处理器31连接,测序平台32用于根据二元性特征,将预获取的目标样本分成正样本集和负样本集,对正样本集和负样本集进行血小板转录组测序,分别获取正样本基因测序读取序列和负样本基因测序读取序列,并传输至处理器31,处理器31用于获取正样本基因测序读取序列、负样本基因测序读取序列和实体的基因表达量估计值根据基于血小板差异表达基因标记的分类方法,如预处理、基因表达量估算、差异表达基因标记确定、超平面表达式构建和量化分类等步骤,获取超平面表达式和该实体的量化分类结果,并传送至服务器33和显示屏34,服务器33用于存储超平面表达式和实体的量化分类结果,显示屏34用于根据显示模式指令显示实体的量化分类结果,触摸屏用于接收用户点选的显示模式指令。
其中,全血中血小板总RNA的提取及转录组测序过程如下:
首先,把加过抗凝剂的全血置于离心机中,在室温下,以加速度120g的条件离心20分钟,去除细胞和其他聚集物,再以加速度360g的条件离心20分钟分离血小板聚集物,并将离心获得的血小板聚集物收集到RNAlater试剂盒(Life Technologies);
将RNAlater试剂盒(Life Technologies)置于4℃培养箱中8小时以上,再存放于-80℃冰箱中,以供后续使用。
在提取RNA时,使用mirVana试剂盒(Life Technologies)承载冻存的血小板聚集物,并将使用mirVana试剂盒(Life Technologies)置于冰上融化。
血小板聚集物融化后,采用SMARTer微量RNA测序试剂盒(Clontech)承载血小板聚集物,获得血小板聚集物中总RNA,并对总RNA进行互补DNA(cDNA)的合成和扩增。
选择能够检测到300-7500碱基长度片段的样品,采用超声破碎的方法(CovarisInc)将RNA打成测序可用的小片段。
采用Illumina的Trueseq DNA Sample Prep试剂盒进行测序样品的制备,最终采用Hiseq2500测序平台32(Illumina Inc.)获取100碱基长度的测序序列,如正样本基因测序读取序列、负样本基因测序读取序列和测序衔接序列。
由上述技术方案可知,本实施例提供的基于血小板差异表达基因标记的分类系统,采用测序平台32测量实体或目标样本的测序读取序列,触摸屏接收用户发送的显示模式指令,处理器31根据提供的基于血小板差异表达基因标记的分类方法,获取该实体的量化分类结果,并根据用户选择的显示模式,在显示屏34上进行直观显示。同时,服务器33能够存储处理器31中的数据,如超平面表达式和每个实体的量化表达结果,以便于后续进行数据查询等。在此,显示模式可以设置为数字显示模式或颜色显示模式,其中,数字显示模式直接显示±1的数字结果,以便于与其他硬件设备连接,方便信息传输。颜色显示模块更直观,有助于增强用户体验。同时,显示屏34可以是设置于医疗设备上的显示屏,此时,处理器31与显示屏34可以采用控制总线连接。同时,显示屏34也可以用户移动端的显示屏,处理器31与显示屏34通过无线连接方式进行信息交互,方便用户实时获取量化分类结果。
因此,本实施例基于血小板差异表达基因标记的分类系统,能够快速、准确地鉴别出特定群组的差异表达基因标记,且对该群组实体的分类判别准确、可靠,数据处理效率高。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围,其均应涵盖在本发明的权利要求和说明书的范围当中。
- 还没有人留言评论。精彩留言会获得点赞!