CRISPR-Cas9系统sgRNA作用靶点的筛选方法及装置与流程
本发明涉及生物信息学、蛋白质组学、转录组学及基因工程领域,具体地说,涉及CRISPR-Cas9系统sgRNA作用靶点的筛选方法及装置。
背景技术:
随着DNA测序技术的发展,许多模式生物的基因组序列信息已被公布,随后科研工作者将研究重点转向对基因功能信息的挖掘上。基因敲除动物模型一直以来是在活体动物上从事基因功能研究、寻找合适药物作用靶点的重要工具。但是传统的基因敲除方法需要通过复杂的打靶载体构建、胚胎干细胞(ES细胞)的筛选、嵌合体繁育等一系列步骤,不仅操作流程繁琐,对实验人员的技术要求很高,而且费用昂贵,耗时较长,且成功率也受到多方面因素的影响。即使对于技术相对成熟的实验室,利用传统技术构建基因敲除大、小鼠模型一般也需要很长时间。
2013年,美国两个实验室在《Science》杂志发表了基于CRISPR-Cas9系统在细胞系中进行基因编辑的新方法,该系统的原理是crRNA(CRISPR-derived RNA)通过碱基互补配对与tracrRNA(trans-activating RNA)结合形成tracrRNA/crRNA的复合物,该复合物可以引导核酸内切酶Cas9蛋白在与crRNA配对的序列靶位点切割双链DNA。而通过人工设计这两种RNA,可以改造形成具有引导作用的sgRNA(short guide RNA),即可引导Cas9对DNA的定点切割,一旦切割完成,细胞会启动各种修复方式来修补被剪掉的部分,其中最常见的是非同源末端连接(NHEJ)的修复方式,该种修复方式使得修复过程很容易出错,这就很大概率地引入使基因功能丧失的变异 (如插入或者缺失部分碱基序列以造成移码突变),这使得研究者能通过突变体来了解被编辑的基因的功能。该项技术已经被迅速应用到基因敲除斑马鱼、小鼠和大鼠等动物模型的构建之中。CRISPR-Cas9技术是继锌指核酸酶(ZFN)和TALEN等技术之后可用于定点构建基因敲除动物的新方法,具有效率高、速度快、生殖系传递能力强及简单经济的特点,在动植物模型构建的应用前景非常广阔。
目前在动物研究领域,有很多基于单个功能基因进行设计的Cas9靶点,但还缺乏一套筛查全基因组靶点的成熟方法。本发明根据Cas9在基因组中编辑靶点的偏好性,开发了一套获取动物全基因组水平Cas9靶点序列的方法。通过此方法设计出来的靶点文库,使得CRISPR可以同时针对全基因组水平的基因靶向,获得高通量的基因突变体库,该方法在基础研究中(例如药物研发和农业)将发挥巨大作用。
技术实现要素:
本发明的目的是提供一种CRISPR-Cas9系统sgRNA作用靶点的筛选方法。
本发明的另一目的是提供一种筛选CRISPR-Cas9系统sgRNA作用靶点的装置。
为了实现本发明目的,本发明提供的CRISPR-Cas9系统sgRNA作用靶点的筛选方法,包括以下步骤:
(1)利用已公布物种的全基因组序列及基因注释信息,获取基因组中具有5’-Nx-NGG-3’序列的区段,作为CRISPR-Cas9系统sgRNA的候选靶点;其中,x为19~22之间的整数,N代表碱基A、T、G或C;
(2)将基因组打断成22~25bp的片段并筛选以NGG结尾的,且在基因组上无重复的序列;
(3)将步骤(1)的候选靶点序列与步骤(2)中筛到的序列进行比对,根据错配信息及评选公式对相应的优选序列进行筛选及排序,获取最优的全基因组sgRNA作用靶点集合。
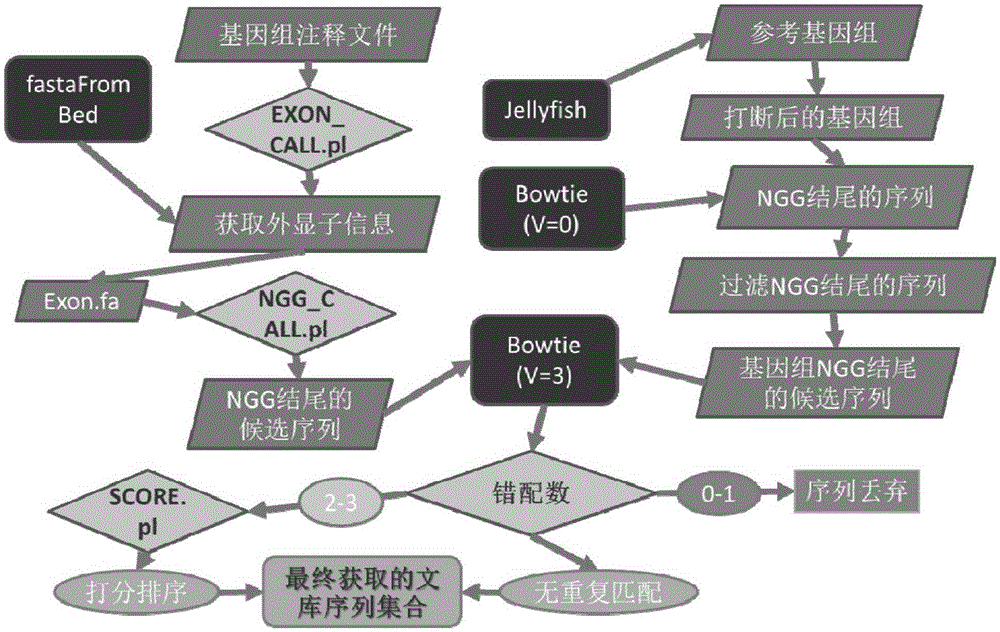
前述的方法,步骤(1)中筛选符合条件的候选靶点序列的要求是:①靶点必须落在基因的CDS区内,即起始密码子之后;②尽可能靠近基因的5’端(实验表明,靠近5’端的外显子,其功能性更强);③优选地,每个基因提取两个外显子(选取两个外显子是为了保证基因被修饰后其功能尽可能地发生变化),具体步骤为:以转录本为单位(若以转录本为单位,一个基因有可能重复取到同一个外显子,下文有去重复的步骤),从基因组注释文件中获取转录本、基因ID,CDS、外显子区的起始和终止位置以及染色体号等相关信息,以每个转录本的起始密码子所在位置为标准,提取其后两个外显子的始末位置,若起始密码子后只有一个外显子,则只取一个,得到候选外显子的始末位置后,利用bedtools软件中的fastaFromBed程序获取这些外显子的序列信息,保留作为外显子NGG候选靶点序列(保存为fasta格式文件)。候选外显子的筛选设计见图1。
其中,fastaFromBed程序中的-s参数的作用是获取反向互补序列,这样就得到了所有外显子的编码链序列信息,便于筛选NGG位点(不用考虑负链,但要注意位置信息)。所有外显子的编码链序列提取它们的前19~22bp序列保存成fasta格式(注意此时的始末位置信息,正负链的情况有所区别,另外由于最终在与基因组水平的NGG序列进行比对时,需去除自比的比对结果,因此就需要详尽了解每一个外显子上的NGG序列所在的基因组始末位置,正负链信息等。因此要进行相应的格式调整)。鉴于上文提到的以转录本为单位会重复取得外显子的情况,进一步对获取的序列进行了去重复处理。最后获得的基因组中所有基因上的候选靶点5’-Nx-NGG-3’序列,统计其覆盖的基因数目,外显子数目,以及候选外显子上获得的NGG位点个数。
前述的方法,步骤(2)中筛选中符合条件的序列的具体步骤为:全基因组筛选采用k-mer打断、再比对找回位置的方法来定位基因组中的NGG序列。首先用jellyfish软件将基因组打断成22~25bp的片段, 考虑到正负链不同,分别筛选正链以NGG结尾和负链以CCN开头的序列,保留作为基因组NGG候选靶点序列(保存为fasta格式文件);由于利用jellyfish软件将基因组打断成22~25bp的片段后没有位置信息,因此需利用bowtie软件比对找回上述22~25bp片段所在基因组中的位置。正负链分别进行比对,比对结束后,将NGG三个碱基从正链中去除,同时将CCN三个碱基从负链中去除,保存为19~22bp的含位置信息的fasta格式文件。
前述的方法,步骤(3)中比对的具体步骤为:
①将步骤(1)的外显子NGG候选靶点序列与步骤(2)的基因组NGG候选靶点序列进行比对,将所有自比结果过滤掉;
②筛选步骤①过滤后的比对结果中,外显子NGG候选靶点在基因组中其它位置上没有比对结果的序列,这些外显子NGG靶点在基因组中是唯一的,将这些靶点序列作为最佳候选靶点序列优先被提取出来,标注为unique reads;
③筛选步骤①过滤后的比对结果中,外显子NGG候选靶点在基因组中其它位置上仍存在比对结果的序列,若出现错配0个碱基(即在基因组其它位置完全比对上)或错配1个碱基(即在基因组其它位置比对上,且只有1个碱基错配),表明这些序列在基因组中有重复序列存在,将这些靶点序列全部删除;
④筛选步骤①过滤后的比对结果中,外显子NGG候选靶点在基因组中其它位置上仍存在比对结果的序列,若出现错配2个碱基(即在基因组其它位置比对上,但有2个碱基错配)或错配3个碱基(即在基因组其它位置比对上,但有3个碱基错配),将这些靶点序列标注为candidate reads,这些reads的所有比对结果通过公式进行打分,打分公式如下:
其中,MS代表错配罚分,a、b、c分别代表发生错配的碱基位置(以该条NGG候选靶点的3’端碱基记为1位,从3’端向5’端依次计数,例如,a为NGG候选靶点3’端上游5bp位置的碱基发生错配,则a等于5),S(ab)代表a与b的代数和,S(bc)代表b与c的代数和,S(ac)代表a与c的代数和,D(ab)代表两个错配碱基a与b的相对位置之差,D(bc)代表两个错配碱基b与c的相对位置之差,D(ac)代表两个错配碱基a与c的相对位置之差;
当n=3时,若S(ab)×D(ab)<S(bc)×D(bc),公式则变为:
若S(ab)×D(ab)>S(bc)×D(bc),公式则变为:
所有reads按照打分从低到高排序,将分数低的前10万条左右的reads作为候选序列(根据下游基因芯片的容量,目前芯片最多容纳10万条序列),即打分通过的candidate reads;
⑤步骤②的unique reads和步骤④打分通过的candidate reads即为最优的全基因组sgRNA作用靶点集合,即初步得到全基因组的Cas9sgRNA Oligo Library。
本发明筛选CRISPR-Cas9系统sgRNA作用靶点的流程图见图2。
本发明的目的还可以采用以下的技术措施来进一步实现。
(1)利用已公布物种的全基因组序列及基因注释信息,获取基因组中具有5’-Nx-NGG-3’序列的区段,作为CRISPR-Cas9系统sgRNA的候选靶点;其中,x为20,N代表碱基A、T、G或C;
(2)将基因组打断成20bp的片段并筛选以NGG结尾的,且在基因组上无重复的序列;
(3)将步骤(1)的候选靶点序列与步骤(2)中筛到的序列进 行比对,根据错配信息及评选公式对相应的优选序列进行筛选及排序,获取最优的全基因组sgRNA作用靶点集合。
其中,步骤(1)中筛选符合条件的候选靶点序列的要求是:i.靶点必须落在基因的CDS区内,即起始密码子之后;ii.尽可能靠近基因的5’端;iii.优选地,每个基因提取两个外显子,具体步骤为:以转录本为单位,从基因组注释文件中获取转录本、基因ID,CDS、外显子区的起始和终止位置以及染色体号相关信息,以每个转录本的起始密码子所在位置为标准,提取其后两个外显子的始末位置,若起始密码子后只有一个外显子,则只取一个,得到候选外显子的始末位置后,利用bedtools软件中的fastaFromBed程序获取这些外显子的序列信息,保留作为外显子NGG候选靶点序列。
步骤(2)中筛选中符合条件的序列的具体步骤为:首先用jellyfish软件将基因组打断成20bp的片段,考虑到正负链不同,分别筛选正链以NGG结尾和负链以CCN开头的序列,保留作为基因组NGG候选靶点序列;由于利用jellyfish软件将基因组打断成20bp的片段后没有位置信息,因此需利用bowtie软件比对找回上述20bp片段所在基因组中的位置。
步骤(3)中比对的具体步骤为:
iv.将步骤(1)的外显子NGG候选靶点序列与步骤(2)的基因组NGG候选靶点序列进行比对,将自比的结果过滤掉;
v.筛选在步骤①过滤后的比对结果中,外显子NGG候选靶点在基因组中没有比对结果的序列,这些外显子NGG靶点在基因组中是唯一的,这些靶点序列作为最佳候选靶点序列优先被提取出来,标注为unique reads;
vi.筛选在步骤①过滤后的比对结果中,外显子NGG候选靶点在基因组中有比对结果的序列,若出现错配0个碱基或错配1个碱基,表明这些序列在基因组中有重复序列存在,将这些靶点序列全部删除;
vii.筛选在步骤①过滤后的比对结果中,外显子NGG候选靶点在基因组中有比对结果的序列,若出现错配2个碱基或错配3个碱基,将这些靶点序列标注为candidate reads,这些reads的所有比对结果通过公式进行打分,打分公式如下:
其中,MS代表错配罚分,a、b、c分别代表发生错配的碱基位置,S(ab)代表a与b的代数和,S(bc)代表b与c的代数和,S(ac)代表a与c的代数和,D(ab)代表两个错配碱基a与b的相对位置之差,D(bc)代表两个错配碱基b与c的相对位置之差,D(ac)代表两个错配碱基a与c的相对位置之差;
当n=3时,若S(ab)×D(ab)<S(bc)×D(bc),公式则变为:
若S(ab)×D(ab)>S(bc)×D(bc),公式则变为:
所有reads按照打分从低到高排序,将分数低的前10万条reads作为候选序列,即打分通过的candidate reads;
viii.步骤v.的unique reads和步骤vii.打分通过的candidate reads即为最优的全基因组sgRNA作用靶点集合。
本发明还提供上述方法获得的CRISPR-Cas9系统sgRNA作用靶点在构建基因敲除突变体文库或基因敲除动物模型中的应用。
本发明还提供一种基因芯片,所述芯片含有根据上述方法获得的CRISPR-Cas9系统sgRNA作用靶点的序列集合。
本发明进一步提供一种筛选CRISPR-Cas9系统sgRNA作用靶点的装置,包括以下模块:
A.全基因组外显子序列提取模块:用于上述步骤(1)中提取全基因组的外显子中具有5’-Nx-NGG-3’序列的区段;
B.外显子NGG序列优选模块:用于上述步骤(1)中所有外显子中具有5’-Nx-NGG-3’序列区段的优选筛查;
C.基因组序列打断比对模块:用于上述步骤(2)中将全基因组序列打断成相应大小的片段,并比对进行位置锚定;
D.外显子NGG候选序列与基因组NGG候选序列比对模块:用于上述步骤(3)中外显子NGG候选序列与基因组NGG候选序列之间的比对;
E.候选sgRNA靶点序列打分模块:用于上述步骤(3)中所有候选序列的评估排序。
本发明提供的方法可应用于所有已知基因组及其基因注释信息的物种当中,通过快速高效获得其全基因组水平的sgRNA序列全集来构建基因敲除突变体文库或基因敲除动物模型。另外,这种高通量的CRISPR-Cas9系统sgRNA作用靶点筛选方法极大降低了成本,克服了单个制备基因敲除细胞,所导致的时间和劳动成本高的问题。
附图说明
图1为本发明候选外显子的筛选设计流程图。其设计原理在于:对位于起始密码子后面的外显子进行设计,如果起始密码子后面有两个以上的外显子,则将这两个外显子全部获取,若起始密码子后面只有一个外显子,则仅取一个外显子;某些起始密码子位于外显子内部,若其后面还有外显子,则从其下一个外显子开始计数,若其后面没有外显子,则从其本身开始计数;负链候选外显子的设计规则同正链。
图2为本发明筛选CRISPR-Cas9系统sgRNA作用靶点的流程图。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规 手段,所用原料均为市售商品。
实施例1针对鸡设计的CRISPR-Cas9系统sgRNA作用靶点的筛选方法
本实施例以禽类代表动物--鸡为例,进行全基因组Cas9靶点文库的设计。
首先在Ensembl数据库(http://www.ensembl.org/index.html)中下载鸡的参考基因组(版本号Galgal4,GCA_000002315.2)及其对应的基因注释文件。利用全基因组序列及基因注释信息,获取基因组中所有基因的候选靶点5’-(N20)NGG-3’序列(N代表A/T/C/G),统计可知,鸡中的候选靶点序列一共获得380,459条,覆盖的基因为16,821个,覆盖的外显子数为28,915个。然后将基因组打断成23bp的片段并筛选以NGG结尾的,且在基因组上无重复的序列,将其与外显子上的候选靶点序列进行比对,根据错配信息及评选公式对相应的优选序列进行筛选及排序,根据下游芯片合成设计容量,共设计了96000条靶点序列,最终筛选结果,覆盖的基因数目为16,569个,每个基因上设计的靶点序列约为7-8个。
实施例2针对猪设计的CRISPR-Cas9系统sgRNA作用靶点的筛选方法
本实施例以哺乳动物类代表动物—猪为例,进行全基因组Cas9靶点文库的设计。
首先在Ensembl数据库中(http://www.ensembl.org/index.html)下载猪的参考基因组(版本号Sscrofa10.2,GCA_000003025.4)及其对应的基因注释文件。利用全基因组序列及基因注释信息,获取基因组中所有基因的候选靶点5’-(N20)NGG-3’序列(N代表A/T/C/G),统计可知,猪中的候选靶点序列一共获得626,236条,覆盖的基因为24,734个,覆盖的外显子数为43,049个。然后将基因组打断成23bp的片段并筛选以NGG结尾的,且在基因组上无重复的序列,将其与外显 子上的候选靶点序列进行比对,根据错配信息及评选公式对相应的优选序列进行筛选及排序,根据下游芯片合成设计容量,共设计了96000条靶点序列,最终筛选过后,覆盖的基因数目为22,731个,每个基因上设计的靶点序列约为4-5个。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!