掺杂剂掺杂的电极及其用途的制作方法
本发明涉及碳阳极的电氧化。特别地,本发明涉及用普通的煤污垢例如锐钛矿、氧化铝、黄铁矿、石英、高岭土和蒙脱石策略性地污染(或掺杂)这类电极的技术效果。
获得的结果尤其与能源生产领域相关-尤其是通过直接碳燃料电池(direct carbon fuel cell,DCFC)和相关技术的能源生产。本文将描述本发明关于其在DCFC技术中的应用,然而,本领域技术人员会理解本发明不限于该特定领域的用途。
背景技术:
本说明书全文中现有技术的任何讨论决不应被认为承认这种现有技术在本领域中是众所周知的或形成本领域中公知常识的一部分。
基于化石燃料的能源生产既不是可持续的,对当前的气候问题也是不利的。现代的能量生产严重地依赖于化石燃料,例如煤、气和油,其全部具有完全有限的储量,且其全部产生排放物,例如二氧化碳(CO2)、氮氧化物(NOx)和二氧化硫(SO2);这些排放物妨碍自然循环和自然过程。
反对这些限制是因为对于电力生产的全球需求几乎是指数地增长。许多当前的研究兴趣集中于设计和开发新的发电技术,其理念上对降低排放强度也有作用。一种这样的技术是直接碳燃料电池(DCFC)。
DCFC并不是新技术。实际上,这种电池的第一个美国专利发布于1896年。DCFC通过在电池阴极内的氧的还原与在阳极侧发生的碳燃料源的氧化来产生电能。电池通过结合碳和氧来产生能量,其释放作为副产物的二氧化碳。使用的碳可以是煤、焦炭、木炭或碳的非化石化来源的形式。在以下的等式(1)至(3)中示出DCFC的半电池反应和全电池反应:
C+2O2-→CO2+4e-阴极 (I)
O2+4e-→2O2-阴极 (2)
C+O2→CO2整体 (3)
使用固体燃料工作的燃料电池比使用气体燃料工作的那些电池能够提供更高的能量密度。例如,固体碳与气体燃料和液体燃料例如甲烷(4.2kWh/L)、氢气(2.4kWh/L)或柴油(9.8kWh/L)相比包含每单位体积高的能量密度(即20kWh/L)。
鉴于以上所述,在材料和电解化学中的最新进展似乎认可这种潜力。事实上,这样的进展已经看到了百年的DCFC根据其工业实用性最近正经历某种程度的复兴。尽管释放二氧化碳,DCFC事实上比传统的碳燃烧技术是更环境友好的;由于其较高的效率,其需要更少的碳来产生等量的能量。并且,因为排放纯二氧化碳,所以碳捕获技术与对于常规发电站相比相对地更便宜,常规的发电站必须或在固定前将二氧化碳与其他排放气体分离,或简单地固定全部排放气体(其可以包括不期望的产物,例如氮的氧化物和硫的氧化物和微粒物质)。如会理解的,这两个选项多少比在其来源捕获纯二氧化碳的设备效率低。DCFC氧化产物的相对纯度实现更简单的固定,而无需昂贵的和耗能的分离和纯化过程。
存在至少四种类型的DCFC:第一种是基于固体氧化物燃料电池(solid oxide fuel cell,SOFC)概念;第二种是熔融氢氧化物燃料电池(参见例如1896年的美国专利555,511);第三种是基于熔融碳酸盐燃料电池(molten carbonate fuel cell,MCFC)概念(参见1897年的加拿大专利55,129);和第四种是熔融锡阳极固体氧化物燃料电池设计,其使用熔融锡和锡氧化物作为溶解于阳极的碳的氧化和在固体氧化物阴极的氧的还原之间的级间反应。
DCFC是独特类型的燃料电池,这是因为其具有使用固体碳燃料的能力。煤,一种众所周知的无定形碳的来源,在世界各地的能量生产中被广泛使用,并且已经被确定作为用于DCFC中的候选[参见参考文献1-5]。假定非Boudouard条件,即碳通过高温下与二氧化碳反应的化学转化[参见参考文献2、6],煤中的含碳材料在DCFC内可以直接转化为电能。该直接转化具有将煤中含有的能量的电转化效率增加至超过80%的潜力(相比较,常规的燃煤发电站,其通常低于40%运转[参见参考文献1],假定只发生一种能量转换)。
在不同电池条件/布置下碳至二氧化碳的氧化反应中的动力学和相关限制作为研究关于DCFC的重要途径最近已经是突出的[参见参考文献1、7]。存在在最近公布的文献中使用的许多电池设计,其提供了在DCFC条件下不同碳材料和电池组分的整体电化学性能的信息[参见参考文献8至15]。然而,较少数的研究小组特别地集中于阳极氧化反应,包括在阳极电极的电化学过程的分析[参见参考文献3、16、17]。因此,DCFC的阳极氧化反应形成了本发明的基础。
优化DCFC的阳极效率的方法已经在专利文献全文中公布。特别需要注意的是Direct Carbon Technologies,LLC的国际公布WO 2008/118139。该公布涉及包含与普通组合物Ai-xA’xRuO3、AB1.yR.UyO3和A1-xA’χBi.yRUyO3相对应的掺杂剂掺杂的钌酸盐的催化阳极,其中A和A’是二价的、三价的和四价的阳离子,B是多价阳离子。实现良好的运行效率,然而会理解的是用通常稀有的过渡金属来掺杂阳极所需要的额外步骤对操作者产生了额外的负担。
最近,圣安德鲁斯大学的国际公布WO 2013/061067吸引了注意。该公开提供包含电化学电池的DCFC体系,所述电化学电池自身包含阴极、固态第一电解质和阳极,其中体系还包括含有和/或适合接受第二电解质和燃料的阳极室。
DCFC的其他实例在国际公布WO 2006/061839和美国公布US 2006/0019132中进行描述,两者都描述了具有固态电解质和含有燃料和液态电解质的阳极的电池。
通常来说,关于DCFC中阳极氧化反应的专利和科学文献确实可以以对化学改性阳极的明显需求为特征。似乎很少关注研究天然存在(即已经掺杂)的材料例如煤和焦炭的阳极特性;本发明对文献中的该明显空缺作出回应。
本发明的一个目的是克服或改善现有技术的至少一个缺点,或提供有用的替代。
本发明的特别优选形式的一个目的是提供通过相对优化阳极氧化反应来提供DCFC的整体效率(即碳转化为电力的百分比)的手段。
通过使用特别设计的电化学测试电池,在该工作中使用石墨作为基础来研究电化学氧化反应。将电池设计为能够测试固体阳极布置而不是在先前的工作中已经使用过的悬浮碳[参见参考文献3、13、17]。在使用悬浮碳作为基础的研究中有许多缺点。主要地,电池的传质限制掩饰了特定燃料的一些其他动力学行为。此外,如果将用矿物质人为污染的碳源悬浮并在熔融盐(电解质)中搅拌任何一段时间,很可能污染物相会与熔融盐相互作用。然而,先前已经假设矿物质残留为碳材料的一部分并影响颗粒表面上的反应[参见参考文献3]。
选择石墨碳作为阳极用于本发明的研究,因其与用于在DCFC中使用而测试的其他碳材料相比,在碳类型、表面功能、含灰量、化学和普通物理和化学行为中表现了显著更低的变化性[参见参考文献8、16、18]。首先确定石墨的基本氧化行为以对比不同污染物对石墨碳的氧化的影响。
基于通常在澳大利亚黑(即含沥青的)煤的样品中存在的那些选择用于研究的污染物。通常在昆士兰州和新南威尔士州开采这样的煤,其被用于国内发电和用于出口。澳大利亚黑煤包含大量的矿物质,通常以黏土、混合的金属/非金属氧化物和黄铁矿的形式,其中一些已经显示以煤灰的形式在测试DCFC电池中影响碳的电化学氧化[参见参考文献19]。在该研究中,使用XRD分析四种澳大利亚次烟煤和美国焦煤以确定在研究的特定煤中以大浓度存在的矿物质相。特别注意确定存在于煤中的污染物,因为这些污染物反过来会被引入到DCFC体系中(而不是随高温灰化确定的那些污染物,已知其影响污染物和煤的化学结构[参见参考文献20至22])。
除非本文明确地要求,否则在本说明书和权利要求全文中,词语“包含”、“包括”等被理解为包括性的意思,与不包括的或详尽的意思是相对的,即是“包含,但不限于”的意思。
如本文所应用的,术语“掺杂”和其变体旨在表达“污染的”或“选择性污染的”。与例如煤会遭遇的天然污染相比,选择性污染指本发明的碳电极已经用外来物质有意地改性。因此,在本文中使用的“掺杂”、“掺杂剂掺杂的”、“掺杂剂”等要求有意地添加污染物质以改善其性能超过天然污染会实现的性能。
尽管本发明会参考具体实施例描述,但是本领域技术人员会理解本发明可以以许多其他形式具体实现。
如本文所使用的,DCFC是电化学电池,其中使用碳作为燃料,所述染料反过来在阳极上通过氧化剂电化学氧化。本文中术语“直接”的使用不指一个基元反应步骤,而是用作表明在一个过程中燃料的直接转化,即不需要外部过程例如破碎。例如,直接反应可以包括一个室中的气化和燃料电池反应。此外,尽管使用术语“燃料电池”,但是会理解的是电化学电池不需要持续地用燃料和/或氧化剂补充。还会理解的是电池的阳极侧和/或阴极侧中的至少一个可以使用更类似于电池组的分批法或单次使用法来操作。
技术实现要素:
本发明一般涉及检查碳的基本电化学行为的方法,其包含半电池装置和固体牺牲阳极的使用。使用该方法,碳的电化学氧化已经使用以与澳大利亚黑煤相同的掺杂剂对碳电极进行选择性污染来评估。确定的污染物包括锐钛矿、氧化铝、黄铁矿、石英、高岭土和蒙脱石。由这些污染物的系统引入,已经发现黏土材料、如高岭土(例如Al2Si2O5(OH)4)和蒙脱石(例如(Na,Ca)0.33(Al,Mg)2(Si4O10)(OH)2·nH2O)起催化作用以提高碳氧化的速率。另一方面,金属氧化物和金属硫化物例如锐钛矿(TiO2)、氧化铝(Al2O3)和黄铁矿(FeS2,“傻瓜的黄金”)在归一化的电流中提供了有限的增加,而石英(SiO2)导致性能的显著下降。当将这些污染物代替地添加到熔融的碳酸盐电解质中时未观察到相同效果;这些结果证明这些污染物的固相相互作用对碳的电化学氧化的明显效果。
根据本发明的第一方面,提供选自高岭土、蒙脱石、氧化铝、锐钛矿和黄铁矿的掺杂剂用于掺入到固体碳工作电极中,以增强电化学半电池内的阳极氧化的用途。
在一个优选实施方案中,电化学半电池存在于直接碳燃料电池(DCFC)中,碳工作电极是阳极氧化的。
在一个实施方案中,掺杂剂和碳在其氧化增强中实质上起催化作用。优选地,碳是石墨的形式。
在一个实施方案中,增强与掺杂剂的相对比例和碳相与掺杂剂相之间的物理接触程度两者相关。碳相与掺杂剂相之间的物理接触程度与相之间的混合均匀性和/或掺杂剂相的任何预处理相关。
在一个优选实施方案中,掺杂剂的相对比例是约10重量%至约50重量%。
优选地,掺杂剂是高岭土或蒙脱石。最优选地,掺杂剂是高岭土。在另一个优选实施方案中,高岭土在粒化前通过在大约500℃加热大约30分钟来进行预处理,以减轻由于在半电池条件下高岭土脱羟基成为偏高岭土对生成的颗粒的可能机械损害。
最优选地,其中经预处理的高岭土以基本上大于30重量%存在,其中电势较低(例如,0.2V vs.C/CO2/CO32-),从而提供45mAcm-2至50mAcm-2等级的氧化增强。
根据本发明的第二方面,提供石英掺杂剂用于掺入到固体碳工作电极中以抑制电化学半电池内的阳极氧化的用途。
在一个实施方案中,随着电势增加抑制趋于基本完成。
优选地,抑制与石英掺杂剂的相对比例和碳相与掺杂剂相之间的物理接触程度两者相关。在一个实施方案中,碳相与石英掺杂剂相之间的物理接触程度与相之间的混合均匀性相关。
在一个优选实施方案中,掺杂剂的相对比例是约10重量%至约50重量%。
根据本发明的第三方面,提供用于并入到电化学半电池内的固体碳工作电极,电极是用选自高岭土、蒙脱石、氧化铝、锐钛矿和黄铁矿的掺杂剂掺杂的,从而提供在半电池内的阳极氧化的增强。
在一个优选实施方案中,电化学半电池存在于直接碳燃料电池(DCFC)中,碳工作电极是阳极氧化的。
在一个实施方案中,掺杂剂和碳在其氧化增强中实质上起催化作用。优选地,碳是石墨的形式。
在一个实施方案中,增强与掺杂剂的相对比例和碳相与掺杂剂相之间的物理接触程度两者相关。碳相与掺杂剂相之间的物理接触程度与相之间的混合均匀性和/或掺杂剂相的任何预处理相关。
在一个优选实施方案中,掺杂剂的相对比例是约10重量%至约50重量%。在本发明的范围内的考虑在约10重量%至约50重量%的一般范围内的任何变化。例如,10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49和50重量%和中间值例如20.5、21.5、22.5、23.5、24.5、25.5、26.5、27.5、28.5、29.5、30.5、31.5、32.5、33.5、34.5、35.5、36.5、37.5、38.5和39.5重量%都是预期的。
优选地,掺杂剂是高岭土或蒙脱石。最优选地,掺杂剂是高岭土。在另一个优选实施方案中,高岭土在粒化前通过在大约500℃加热大约30分钟来进行预处理,以减轻由于在半电池条件下高岭土脱羟基成为偏高岭土对生成的颗粒的可能机械损害。
最优选地,其中经预处理的高岭土以基本上大于30重量%存在,其中电势较低(例如,0.2V vs.C/CO2/CO32-),从而提供45mAcm-2至50mAcm-2等级的氧化增强。
根据本发明的第四方面,提供用于并入到电化学半电池内的固体碳工作电极,电极是用石英掺杂的,从而提供在半电池内的阳极氧化抑制。
在一个实施方案中,随着电势增加抑制趋于基本完成。
优选地,抑制与石英掺杂剂的相对比例和碳相与掺杂剂相之间的物理接触程度两者相关。在一个实施方案中,碳相与石英掺杂剂相之间的物理接触程度与相之间的混合均匀性相关。
在一个优选实施方案中,掺杂剂的相对比例是约10重量%至约50重量%。
根据本发明的第五方面,提供增强直接碳燃料电池(DCFC)的效率的方法,该方法包括在DCFC的阳极半电池中并入用选自高岭土、蒙脱石、氧化铝、锐钛矿和黄铁矿的掺杂剂掺杂的固体碳工作电极。
在一个优选实施方案中,电化学半电池存在于直接碳燃料电池(DCFC)中,碳工作电极是阳极氧化的。
在一个实施方案中,掺杂剂和碳在其氧化增强中实质上起催化作用。优选地,碳是石墨的形式。
在一个实施方案中,增强与掺杂剂的相对比例和碳相与掺杂剂相之间的物理接触程度两者相关。碳相与掺杂剂相之间的物理接触程度与相之间的混合均匀性和/或掺杂剂相的任何预处理相关。
在一个优选实施方案中,掺杂剂的相对比例是约10重量%至约50重量%。
优选地,掺杂剂是高岭土或蒙脱石。最优选地,掺杂剂是高岭土。在另一个优选实施方案中,高岭土在粒化前通过在大约500℃加热大约30分钟来进行预处理,以减轻由于在半电池条件下高岭土脱羟基成为偏高岭土对生成的颗粒的可能机械损害。
最优选地,其中经预处理的高岭土以基本上大于30重量%存在,其中电势较低(例如,0.2V vs.C/CO2/CO32-),从而提供45mAcm-2至50mAcm-2等级的氧化增强。
根据本发明的第六方面,提供抑制直接碳燃料电池(DCFC)的效率的方法,该方法包括在DCFC的阳极半电池中并入用石英掺杂的固体碳工作电极。
在一个实施方案中,随着电势增加抑制趋于基本完成。
优选地,抑制与石英掺杂剂的相对比例和碳相与掺杂剂相之间的物理接触程度两者相关。在一个实施方案中,碳相与石英掺杂剂相之间的物理接触程度与相之间的混合均匀性相关。
在一个优选实施方案中,掺杂剂的相对比例是约10重量%至约50重量%。
根据本发明的第七方面,提供制备用于在直接碳燃料电池(DCFC)的阳极半电池内使用的掺杂剂掺杂的固体碳电极的方法,该方法包括与选自高岭土、蒙脱石、氧化铝、锐钛矿、黄铁矿和石英的掺杂剂一起研磨固体碳;和粒化得到的研磨混合物。
优选地,碳是石墨的形式。在一个优选实施方案中,掺杂剂以约10重量%至约50重量%的量存在于掺杂剂掺杂的电极中。
在一个实施方案中,方法还包括预处理步骤,预处理步骤包括在粒化前将掺杂剂在大约500℃加热大约30分钟。
最优选地,掺杂剂是高岭土。在一个优选实施方案中,高岭土在粒化前通过在大约500℃加热大约30分钟来进行预处理,以减轻由于在半电池条件下高岭土脱羟基成为偏高岭土对生成的颗粒的可能机械损害。
根据本发明的第八方面,提供通过如根据本发明的第七方面所限定的方法制备的掺杂剂掺杂的固体碳电极。
在以下实施例中,试验的最大掺杂剂浓度为50重量%。然而,本发明人预期更高的掺杂剂浓度。不希望受理论限制,本发明人假定随着添加更多的污染物,碳氧化过程的利用最终会增加至加入更多的污染物也没有足够的碳存在以进一步增加电流的点。因此,会观察到电流密度的最大值,在该点会达到效率与总电流之间的协调。尽管从实践观点来看,加入电极的更多污染物最终会意味着已经进行氧化后电解质中的更多污染物。这对DCFC的实际应用、特别是电解质再生具有启示。另一方面,这些物质溶解到电解质中实际上具有以下潜在优势,使得能够使用该污染的电解质作为例如铝、硅等的物质的源,从其中可以电沉积出溶解的污染物(在单独的电池中),提取纯净形式的这些物质;同时再生用于在DCFC中使用的电解质。
附图说明
将参考附图仅通过示例的方式描述本发明的一个优选实施方案,其中:
图1是电极的总体布置图。通过与导电性陶瓷黏合剂(Resbond 989,与15重量%的石墨混合)适当地黏合的镍铬合金电线来完成与碳颗粒的接触。将碳颗粒和接触物安装到陶瓷管中,黏合,和适当地加工处理。在图1中,镍铬合金导电线表示为(1)、氧化铝管为(2)、陶瓷胶为(3)、导电陶瓷胶为(4),工作电极颗粒为(5)。
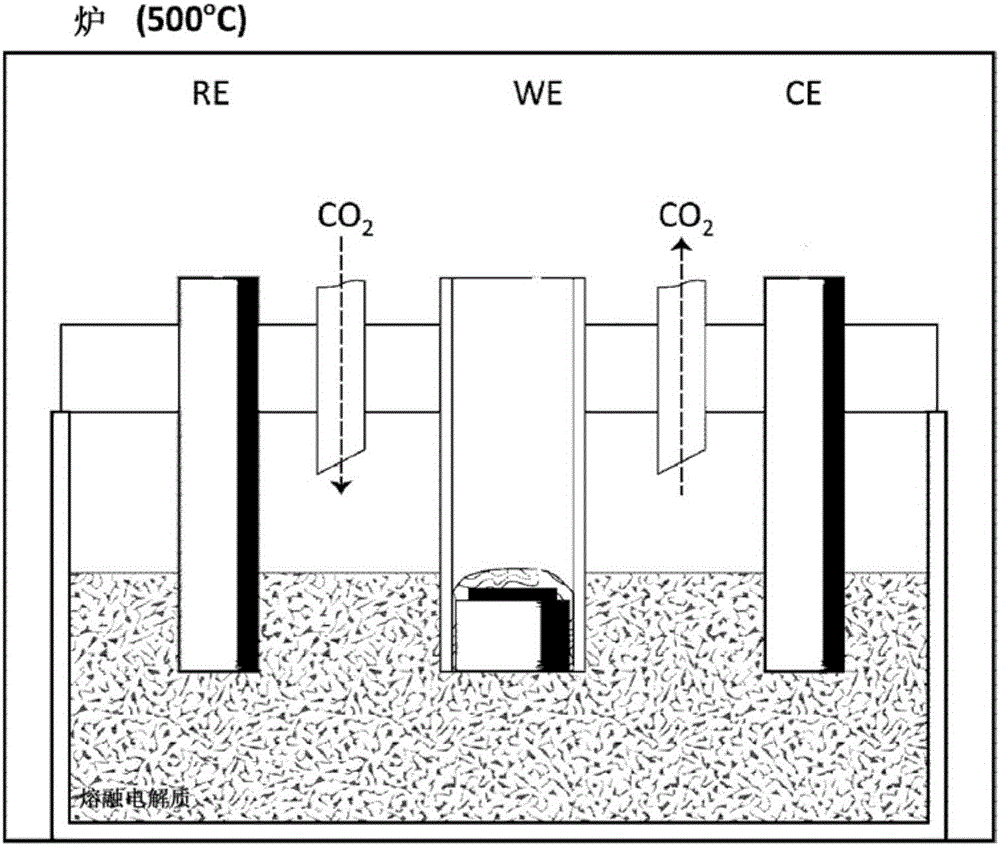
图2是测试电池设计和装置的示意图。制造三电极高温电化学测试电池用于测试熔融碳酸盐共熔合金电解质中的颗粒。电池由具有机械加工的平顶的圆形陶瓷盘和为使工作电极(WE)、参比电极(RE)和对电极(CE)能够适当地牢固保持而特别制造的瓷砖盖组成。通过以50mLN/分钟的速率流动的外部气体供料管线在电池内保持恒定的二氧化碳气氛。
图3示出[A]原煤和[B]LTA残留物(κ-高岭土、θ-石英、π-黄铁矿)的XRD图谱。
图4示出石墨阳极的电流密度相对于电势的线性扫描伏安图。以5mV/s和60秒的扫描间隔来对石墨(纯)电极进行重复的LSV扫描。每个图几乎是相同的,表面使用石墨固体阳极的电化学行为在多次扫描中是可重现的和一致的。
图5示出复制的石墨工作电极的电化学测试结果,显示开路电势(右轴)和在0.3V vs.C/CO2/CO32-的电流密度(左轴)。来自由相同碳源制造的电极的结果给出了可重现的和一致的结果;这不考虑在最终表面形貌中可能的小偏差,其可能由使用的人工电极制备方法导致。
图6示出用[A]高岭土、[B]蒙脱石、[C]氧化铝、[D]锐钛矿、[E]黄铁矿和[F]石英污染的石墨的电化学响应。显示用不同的污染物负载测试的全部污染物的结果。随着污染物的量增加,可用于电化学氧化的活性面积下降,从而反应在减少的表面积上发生(假定污染物在研究的电势范围中是非电化学活性的)。
图7示出掺入不同污染物的石墨在[A]0.0V vs.C/CO2/CO32-和[B]0.2V vs.C/CO2/CO32-测量的电流密度的比较性能。
图8描述在使用前具有[A]高岭土、[B]氧化铝、[C]锐钛矿、[D]黄铁矿和[E]石英的50重量%污染的工作电极表面的扫描电子显微镜(SEM)图像(500倍放大率)。确定为石墨的面积用“G”表示,而污染物面积用“C”表示。结果显示污染物在石墨电极内分布的一些差别。
图9是使用5mV/s的扫描速率在SFG15石墨电极颗粒上进行的线性扫描伏安图。突出了感兴趣区域。该观察结果(在0.12V至0.19V区域中)表明了在电极表面发生另一氧化过程。在该峰后观察到在归一化的电流响应中可识别的减少。
实验过程
煤的表征和处理
a)所研究的煤的近似分析
对煤材料的近似分析使用ATSM D3175-11方法。还使用ASTM D4326-11对煤进行灰分分析。
b)低温灰化
用以提供持续的氧等离子体(~13.65MHz)必需的频率提供200W至240W的RF电源在氧等离子体低温灰化器(PE100Plasma Etch)中进行低温灰化。将经选择、预干燥(在95℃)的20g至30g样品均匀地分布在150mm的Pyrex盘上,并载入到灰化室中,然后将灰化室抽真空至0.15托。通过抽气管路(5至30毫升/分钟)和除气真空泵保持低压氧(BOC工业级)气氛。每48小时时间段,将样品重新称重和轻轻地翻转。当48小时周期后重量损失不大于5mg时假定灰化完成,在3周后观察到完成的灰化。在完成后,将样品密封在密封容器中直到进一步的分析。
c)结构分析
使用Philips PW1710衍射计记录选择的样品的X射线衍射(XRD)图谱。分别使用40mA和40kV、10°至90°的2θ、0.05°2θ的步长、2秒的扫描步长时间的设置使用CuKα辐射在室温下分析样品;发散狭缝-接收狭缝-分散狭缝的宽度分别为1°-0.2°-1°。
d)形态分析
进行制造的电极表面的SEM。为了实现粒化碳样品的显微镜检查,制备抛光的样品。这通过首先将碳颗粒在压力下安装在两部分、冷固的环氧树脂中来实现。然后使用不同等级的碳化硅纸在旋转的转盘上研磨样品,并最终使用金刚石和二氧化硅混合物抛光样品。这样导致较平的、代表性的横截面,使显微镜检查成为可能。为了SEM检查,将样品安装在铝桩上,并用适用于成像和元素X射线分析的碳的20nm导电层进行碳蒸发。在具有硅漂移X射线检测器(SDD)和背散射电子(BSE)检测器的Zeiss MA15仪器上获取SEM图像。
工作电极制造和选择性污染
a)石墨颗粒的制备
在13mm直径压粒机中制备石墨(SFG15;Timcal)颗粒,以740MPa压缩5分钟。为保证测试条件下的稳定性,然后将石墨颗粒在氮气气氛下在500℃烧结4小时,其在外观上或机械强度上无可观察到的变化。烧结后,测量石墨颗粒的电阻率,得到8.0μΩm至9.3μΩm。
b)污染的石墨颗粒的制备
在与石墨材料混合前通过干磨矿物相和使矿物相通过40μm测试筛网将污染物的粒度保持低于标准尺寸。在粒化前将高岭土在500℃下进一步加热处理30分钟,以最小化在电化学测试期间由于在高温下高岭土脱羟基为偏高岭土对颗粒的可能机械损害[参见参考文献23]。
在筛分后,当在研钵及研杵中混合和进一步结合5分钟或直至产生均匀的粉末时将污染物缓慢地引入到石墨粉末中。然后将颗粒相同地制备为纯石墨。
包括氧化铝(Al2O3)、石英(SiO2)和锐钛矿(TiO2)的污染物材料来源于Sigma Aldrich,而高岭土、蒙脱石和黄铁矿来源于在澳大利亚新南威尔士的猎人谷区域的矿床。为确定获得的污染物矿物相的同一性和纯度,通过XRD分析全部污染物。发现全部材料与材料的纯参考物(在无机晶体结构数据库中得到的)匹配,具有细微偏差。观察到的偏差包括在锐钛矿中非常少的金红石相的存在和在黄铁矿材料中石英的少量痕迹。
c)电极结构
通过与导电性陶瓷黏合剂(Resbond 989,与15重量%的石墨混合)适当地黏合的镍铬合金电线来完成与碳颗粒的接触。使导电黏合剂、颗粒和电线接触物在室温下干燥4小时,之后将其在氮气气氛中分别加热至90℃2小时、加热至120℃1小时和加热至300℃1小时,以干燥、加工处理和后加工处理黏合剂。然后将碳颗粒和接触物安装到陶瓷管中,黏合,并使用陶瓷黏合剂(Resbond 989)用与导电胶加工处理相同的方法适当地加工处理。在图1中示出电极的总体布置图。
将工作电极的表面在1000粗砂碳化物砂纸上抛光平,用Milli-Q水充分地冲洗以移除任何表面污染物。该过程制备具有1.766cm2的几何横截面积(考虑表面粗糙度)的工作电极,其已被用于归一化在电化学测试中记录的电流(连同用于以下所讨论的活性碳表面积的归一化)。
电化学电池装置和测试
使用EG&G Princeton Applied Research(PAR)273A恒电势器/恒流器进行在该工作中进行的电化学实验。通过PAR提供M270电化学研究软件以通过国家仪器高速USB通用接口总线(GPIB-USB-HS)控制恒电势器/恒流器。
a)电化学布置
制造三电极高温电化学测试电池用于测试熔融碳酸盐共熔合金电解质中的颗粒。电池由Ceramic Oxide Fabricators(澳大利亚)制备和机械加工的高密度氧化铝(陶瓷)构造。电池由具有机械加工的平顶的圆形陶瓷盘和为了使工作电极(WE)、参比电极(RE)和对电极(CE)适当牢固地保持而特别制造的瓷砖盖组成。通过以50mLN/分钟速率流动的外部气体供料管线在电池内保持恒定的二氧化碳(BOC等级4.5)气氛,以保持恒定的参比电极条件。对所有实验使用500℃的温度。在图2中示出示意性的测试电池设计和设置。
b)电极
对电极(在图2中表示为“CE”)由通过内部黏合的镍铬合金线(如上的导电陶瓷黏合剂和固化策略)具有电接触的石墨棒(GrafTech)组成。在30%的HNO3中清洁石墨电极,用Milli-Q水清洗,并在氮气下加热至500℃以移除来自制造的任何残留物。对电极的几何表面积为工作电极的几何表面积(基于15mm的电解质深度)的约3.7倍,这保证任何表面积限制不是对电极的结果。
c)三碳酸盐电解质
将碳酸钠(Na2CO3)、碳酸锂(Li2CO3)和碳酸钾(K2CO3)(Sigma>99%纯度)在组合前在110℃在真空中干燥24小时。将三种碳酸盐粉末以43.5%Li、31.5%Na、25%K的摩尔比组合(m.p.397℃[参见参考文献24]),并用研钵和研杵轻轻地碾磨5分钟。然后将三碳酸盐前体在110℃下再干燥1小时,并在铂坩埚中、在CO2(BOC等级4.5)气氛中、在450℃下处理30分钟以形成三种组分的共熔合金.
近似分析结果和灰分分析结果
表1概括了五种煤样品在干基上的近似分析结果。对煤材料的近似分析使用ATSM D3175-11方法。还使用ASTM D4326-11进行灰分分析。
表1:澳大利亚煤的五种样品的近似分析(干基上的重量%)
由近似分析中明显的低灰分含量和高固定碳,“煤-C”样品已经被处理以去除灰产生相中的一些是清楚的。全部其他煤材料显示次烟煤的典型近似分析结果。
司空见惯地,煤灰的元素组成报道为具有基于重量百分比表明的组成的氧化物,结果在表2中示出。
表2:煤的灰成分分析
表2中示出的结果表明全部煤灰都是二氧化硅为主的,具有氧化铝和铁氧化物的大量内含物。其他成分是次要的,其组合占总灰分的不到百分比十。煤-C再次显示与其他煤是不同的,其具有显著更小浓度的二氧化硅、氧化铝和铁氧化物,表明在预处理过程中优先去除这些物质。
确定感兴趣的煤矿物相
使用XRD分析原煤(表1中提供的近似分析)。在图3[A]与煤的XRD图谱中示出煤的结果,所述煤已经历了上面概述的低温灰化(“LTA”)过程(参见图3[B])。
全部原煤材料在10°2θ至30°2θ和30°2θ至60°2θ中显示两个宽峰,其已知为贫结晶碳材料的特征[参见参考文献3、25]。有几个来自原煤样品中的结晶矿物质的峰叠加在宽碳峰上;即高岭土(在图3中以κ表示)和石英(在图3中以θ表示),具有在12.1°2θ、24.5°2θ的高岭土峰和在20.5°2θ的独特多峰。由石英产生的20.45°2θ和26.4°2θ的峰对于所有具有20.45°2θ的主石英峰的原煤材料是独特的,其在所有原煤图谱中给出最大峰。
来自石英相的该峰是稍微夸大的,因为煤材料中的石墨碳在26.15°2θ也具有主峰。然而,在49.3°2θ和59.4°2θ的副峰确定了原煤中石英相的存在。在煤-D的情况中,这些副石英峰在背景中是次峰,很可能是由于在煤-D原煤中的石英的大多数被束缚在样品的黏土材料中。原煤-D样品的XRD图谱还显示在32.55°、36.5°、40.35°、46.95°和55.8°2θ的小峰,其是原煤中黄铁矿(用π表示)相的独特主峰。
在图谱中大的无定型碳峰会遮蔽来自其他矿物相的许多峰,这意味着在样品中存在的矿物相的详细信息是难以确认的。使用LTA来去除煤中的碳材料,同时通过显著地降低灰化温度和氧化条件的严重程度来保存样品中存在的矿物相。在图3[B]中示出对于每种煤样品收集的来自LTA残留物的XRD图谱。
低温灰分XRD图谱接近类似于移除了屏蔽宽碳变形的其母煤材料的图谱。所有图谱显示在12.1°2θ和24.7°2θ的强高岭土峰和在19.9°2θ和38.1°2θ的多峰。可以在26.4°2θ清楚地观察到大的石英峰,在20.5°、36.1°、49.8°和67.5°2θ的次峰也是明显的。根据这些特征明显的是,低温灰化未破坏煤样品中存在的矿物相。
对比煤-C和煤-E原煤和LTA残留物的XRD结果,明显的是,在煤-C样品上使用的选矿过程显示了偏向于一些相的去除,同时减少煤样品的整体矿物含量。对于这点的证据是在煤-C图谱中缺少任何明确定义的黏土(高岭土)峰,和来自石英矿物相的峰高的明显降低。根据近似分析,煤-C和煤-E的煤都显示高的Si:Al比例,表明在这些样品中很有可能存在更高的石英相。煤-E显示来自石英相的强主峰和副峰,而副石英峰在煤-C原煤光谱的背景中几乎消失。通过XPert分析软件,在煤-C中确定仅石英和多晶石墨。
表3:在一些煤材料的LTA样品中确定的矿物相
煤-A
煤-D
煤-E
根据该分析明显的是,煤材料比在上文详述的近似分析中报道的碱性氧化物具有更宽的矿物化学。通过使用SiroQuant和XPert XRD分析软件两者,在这些LTA样品中确定大量矿物相。上面的表3突出了在这些LTA残留物中发现的矿物相的范围。
石英和锐钛矿是在LTA样品中发现的仅有的大量氧化物相。该结果得到近期研究[参见参考文献27]强烈支持,其还确定了在LTA结果的汇集中发现的仅有的大量氧化物是石英(SiO2)和锐钛矿(TiO2)。该结果说明测试煤污染物的效果因为其很可能被引入DCFC而需要添加大量的矿物相而不是其氧化物对应物。
石墨性能、稳定性和重现性
为了确定如在新电极设计中作为基线压制颗粒的石墨的性能,使用几种测试方法来确定其不添加电极污染物的氧化性能和重现性。在组装的测试电池中使用石墨工作电极(使用上面概述的过程制备的)以及石墨棒对电极和具有标准碳酸盐共熔合金的参比电极。
由于制备的碳电极颗粒的牺牲性质,确定其对于在一系列连续的电化学扫描中测试持久的电化学性能是重要的。这么做是确保在实施电化学测试所需的时间范围中不出现性能降低。
一旦制备了如图2所示的电池,将其加热至500℃并使其平衡30分钟。在平衡后,在选择的电势范围、即从电池的开路电势(OCP)至相对于参比C/CO2/CO32-高于OCP 0.5V进行大量扫描。在30分钟的时间段内采用5mV/s的扫描速率。这等于以每次扫描间60秒休息间隔十次连续的电势扫描。在图4中示出该实验过程的阳极响应。
图4示出每次重复扫描的几乎相同的i-V曲线,表明使用石墨固体阳极的电化学行为在多次扫描中是可重现的和一致的。在i-V曲线中看到有少量变化,很可能是由于电极表面的变化,其来源于每个连续电势扫描的碳材料的消耗。因为氧化不取决于如具体类型电池的碳至电极表面的传质,所以在图4中未观察到传质限制。这也表明CO32-物质从电解质本体至电极-电解质界面的扩散不受限制,这是因为在半电池体系中,阳极反应还取决于碳酸盐的存在,即:
大范围的文献中报道了在各种热条件下碳酸盐阴离子在混合的碱金属盐中的扩散系数是0.85至1.92x10-5cm2s-1[参见参考文献28至31]。然而,在电化学环境中、即在存在电势梯度的条件下,离子的扩散率可以表现地显著不同。Costa等人[参见参考文献32]报道了在应用的电势梯度下混合的碱性碳酸盐熔融盐中CO32-离子的扩散系数平均增加40%。这些作者还报道了扩散系数为3.0至4.7x10-5cm2s-1。该扩散率的显著增加连同碳酸盐离子物质占熔融电解质约33%的摩尔比例的事实,是在电池内不考虑静态条件时缺少扩散限制的行为的原因。因此反应是在动力学上受限的。
为了确定实验的重复性,其中可能会出现制备工作电极中的较小差异(由石墨的研磨、粒化和表面处理中的差异造成),使用相同的方法制备多个电极并在半电池体系中进行测试。使用的电化学测试过程包括体系的开路电势的确定和以5mV/s至测量的OCP以上0.5V的三个之后的线性扫描(与图4相似,在扫描之间使用60秒的等待时间)。在图5中示出对于每个电极复制品在0.3V vs.C/CO2/CO32-的OCP和最大电流密度(进行的三次扫描的平均值)。
在图5中可以看出,由相同碳源制备的电极的电化学结果提供可重现的和一致的结果。发现平均OCP为-0.224±0.003V vs.C/CO2/CO32-,在0.3V的电流密度平均为9.33±0.49mA·cm-2。这不考虑在最终表面形态中可能的小偏差,其可能是由所使用的人工电极制备方法导致的。
图4和图5中显示的结果表明用于石墨电极基础的良好的电化学重现性和稳定性,在其上可以进行进一步的污染研究。
石墨电极的选择性污染
根据在该研究中使用的原始煤样品及其低温残留物的XRD和近似分析结果(见上)明显的是,石英、黏土(特别是高岭土和蒙脱石)和黄铁矿是在更通常发现的污染物矿物相中。在高温灰分分析(参见表2)中也以高浓度存在的矿物相包括锐钛矿和氧化铝,其也被选择用于污染物研究。
使用如图4和图5所描述的相同电化学过程和第三连续LSV作为代表性扫描,测试石墨的10重量%至50重量%的污染物浓度。图6中示出用各种污染物负载测试的所有污染物的结果,比较在图7中包括的两种不同施加电势下可获得的电流密度。
在图6和图7中电流密度已经归一化,以反映对于每种污染物负载在电极表面存在的活性石墨的相对表面积。随着污染物的量增加,可用于电化学氧化的有效面积下降,从而反应在减少的表面积上发生(假定污染物在研究的电势范围中是非电化学活性的)。通过基于每种污染物的密度和固体石墨的密度计算其体积重量来进行标准化。
然后改变用于标准化每单位面积的电流的几何表面积以反映存在的碳的相对体积比例。这使得能够评估每单位面积的石墨而不是暴露于电解质的总表面积所产生的电流。
在电化学测试前在每个电极材料的表面上进行SEM以确定电极表面的均化和所使用的归一化方法。图8中示出的结果表明污染物在石墨电极内的分布的一些差别。
在每种情况下使用500倍的放大率用于比较污染物相。50重量%污染的电极的表面的SEM图像证实,在所有情况下污染物被紧密地混合到石墨材料内。然而,根据所使用的污染物的类型观察到表面结构中的清晰差别。
高岭土(见图8[A])表现为在石墨中高度分散的污染物中最紧密混合的。可以看出高岭土污染物具有3μm至8μm的粒度。然而,由于一些团聚,一些富高岭土区域可以大到25μm。很可能高岭土至偏高岭土的转化导致了高岭土材料的粒度和硬度的下降。这些反过来导致了来自用于将污染物引入到石墨中的碾磨步骤的更精细地和更均匀地分散的污染物。发现蒙脱石具有与高岭土非常相似的结果。
尽管在氧化铝的情况下观察到更多的石墨富集区域,但是石英和氧化铝(分别参见图8[E]和8[B])表现为在粒度和分散方面是相似的。尽管平均粒度表现为主要在10μm至20μm,但是这些微粒具有4μm至5μm到最高45μm的粒度分布。
锐钛矿污染物形成更大的团聚区域和小的精细分离的颗粒(参见图8[C])。很可能在SEM图像中看见的“大理石纹”类型效果是用于SEM的制备方法的结果。
用于SEM的电极表面的制备需要用非常精细的磨料的湿抛光,其会趋向更容易地从表面去除较软的石墨材料和将锐钛矿涂抹到产生的空腔和裂缝中。根据锐钛矿污染的电极的SEM图像明显的是污染物相间的清晰差别,其显著小于渗透穿过石墨颗粒的石墨相,而不是渗透穿过污染物相的石墨颗粒。
黄铁矿显示最大的相对粒度,从而显示石墨和污染物之间最低的接触。黄铁矿是最硬的污染物(6.5至7莫氏硬度)之一,用于减小黄铁矿材料的粒度的研磨过程导致从3μm至32μm变化的大粒度范围,以及颗粒在上界限中的大发生率。因此,相比于工作电极中的其他污染物,预期黄铁矿污染物具有更大的非均匀粒度分布。
因此认为使用的归一化技术在锐钛矿的情况下(电流密度很可能大于表现的)过度代表石墨,而在添加黄铁矿的情况下(电流密度很可能小于通过归一化所显示的)低度代表石墨,然而,在大多数情况下,其是用于确定活性表面积的良好近似。由图6和图7可以看出,利用该归一化,向电极添加污染物对反应具有显著影响,其一般地随着污染物的添加而增加。
碳酸盐电解质的污染
补充研究污染石墨工作电极的电化学效果,还使阳极作为固体(未掺杂的)石墨进行电解质的污染。然后用高岭土、蒙脱石、锐钛矿、氧化铝、黄铁矿和石英有目的地污染电解质。
将1重量%和5重量%的污染物添加至电解质来进行污染研究。使用如前所述的相同电化学过程来评价在现有的基于液相的基于煤的污染物的存在下石墨的电化学性能。
与将污染物添加至电极相比,发现对于电解质中测试的几乎所有污染物,对于石墨电氧化未看到电流响应的可识别变化。如图5中显示,获得的LSV曲线之间的差别不多于在电极制备过程中的正常变化。在LSV行为中显示小变化的仅有的污染物是石英污染物。如图9中所示,1重量%和5重量%的污染物负载对以5mV/s扫描速率的石墨工作电极的i-V曲线均具有影响。
在石英污染的电解质的i-V曲线中的在图9中可以看出的区别特征是在0.12V至0.19V区域的电流响应中峰的出现,表明在电极表面发生另一氧化过程。沿着该峰,观察到在归一化的电流响应的可识别降低,在5%受污染的电解质中是最显著地。该降低相比于将污染物代替地添加至固体电极时所观察到的变化是不显著的。
结果的技术意义
结果表明在紧密物理接触、即在固体电极中结合的情况下合并的污染物与石墨碳的明显相互作用。测试的污染物的活性顺序表明按高岭土>蒙脱石>氧化铝>锐钛矿>黄铁矿的顺序增加的氧化活性。石英是显示石墨的氧化活性的明显降低的仅有的测试污染物。在其他污染物添加的情况下未观察到相同的效果,对于增加污染物内含物观察到增强的效果。
对于在低电势和高电势添加的每种污染物都可以观察到对增加污染物浓度的相似响应,对于高岭土和蒙脱石在较高的污染物浓度观察到偏差,其增加超过从其他污染物观察到的响应。
对于阳极污染所观察到的最大增强是对于经预处理的高岭土,其也显示在混合上具有与石墨最紧密的接触(参见图8[A])。反应的清楚活化随着增加高岭土和蒙脱石浓度而发生,其在发生氧化反应的明显活化的低电势范围中在大于30重量%的浓度下尤其明显(参见图7[A])。
活化的原因是难以确定的,尽管一些作者先前已经假设可以机械地改变碳的阳极氧化的方式。例如,污染物相可以作为用于交换O2-物质的调节位点,和在相接触的地方可能催化反应。Li等人[参见参考文献3]和Wang等人[参见参考文献16]均注意到当将具体金属氧化物引入到电解质时,其测试电池的性能增强。Li等人将对测试的不同碳源所观察到的性能增强归因于碳相中表面氧化物的增加[参见参考文献3]。高岭土和蒙脱石均包含表面氧化物[参见参考文献26],且在这些结构中的氧化物可能促进O2-到电极表面的吸附和之后与相邻的碳颗粒的反应。
或者,污染物的催化效果可以是熔融电解质和碳表面之间接触的结果。当以高浓度掺入时,高岭土和蒙脱石提供最大的性能增强,并且还观察到其具有最大的混合度和在石墨与污染物之间的接触(参见图8[A])。
污染物添加可以通过改变电极表面在污染物相存在的区域中的可湿性来实现熔融电解质和碳之间的更紧密接触。Chen等人[参见参考文献33]确定碳与碳酸盐电解质之间的接触为可能的限制,且其在近期的综述论文中作为可能的限制问题被讨论[参见参考文献7]。
综上,石英内含物对电极表面的影响是最显著的结果,因为其表现为几乎完全地抑制了在高电势下存在的石墨的氧化(参见图7[B])。考虑到在熔融的碳酸盐电解质中石英的可溶性质[参见参考文献34],石英污染物可能溶解并在电极表面形成在局部区域中减少CO32-根离子浓度的钝化层。
Li2CO3+SiO2→Li2SiO3+CO2 ΔG=-88.48千焦/摩尔 (5)
Na2CO3+SiO2→Na2SiO3+CO2 ΔG=-46.48千焦/摩尔 (6)
K2CO3+SiO2→K2SiO3+CO2 ΔG=-39.43千焦/摩尔 (7)
Devyatkin等人[参见参考文献34]提出了在熔融的三共熔合金碳酸盐/SiO2混合物中可能的一系列化学平衡,预计其中的一些自发地发生并且在非电解质条件下。Devyatkin提出的在SiO2和熔融碳酸盐之间的反应(参见上面的等式(5)至(7))意味着中间体物质M2SiO3(其中M=Li、Na、K)可以存在于电极界面处的电解质中,引起通过其氧化碳的不同系列的电化学反应。
还观察到将石英添加至电解质的影响(图9),其在0.12mV至0.19mV区域是小氧化峰的形式。未见其他杂质具有对石墨的氧化反应的任何影响。尽管由于用于氧化的微粒碳未能够观察到精细的电化学影响,但是Li等人[参见参考文献3]报道了在其碳负载的基础上仅8重量%SiO2(其等于相对于熔融碳酸盐电解质的0.6重量%)的内含物在电化学性能中的显著区别。在涉及煤和在DCFC中的使用的文献,例如Li等人[参见参考文献3]、Cherepy等人[参见参考文献17]和Vutetakis等人[参见参考文献19]中未讨论在石英污染的碳源中的该特征,其很可能被这些研究中使用的电池的传质限制所遮蔽。
Devyatkin等人[参见参考文献34]研究了在其他电极类型中使用玻璃碳电极的碳酸盐熔融物中SiO2的电化学行为。在玻璃碳上,在阳极伏安图(校正使用的参比电极和电池温度)的相同区域中看到峰的出现。
此外,Devyatkin等人报道了在反电势扫描期间工作电极上无相应的阴极过程,表明该过程是不可逆的或动力学上非常缓慢。文献表明在阳极扫描中产生峰的过程是由于碳化硅的电化学氧化,所述碳化硅被认为是在升温过程期间在电极表面化学地形成的。Devyatkin还用在熔融电解质中增加碳含量的一系列实验确定这点,报道了在工作电极上形成黑α-SiC涂层。该影响可以归因于由于SiO2形成造成的石墨表面的活性位点的钝化。
与其他作者将金属氧化物添加至碳酸盐熔融物中的结果相反[参见参考文献3、16],未观察到其他污染物的影响。使用的电池的静止性质在该研究中对高岭土和蒙脱石可能都具有影响,这是因为两种黏土材料均从电解质中沉淀,在电池底部形成固体沉积物,并防止材料与电极表面接触。
开发的方法和电池设计显示为对于确定污染物对在选择的熔融介质中的模型燃料源的电化学影响是有效的。方法还可以应用于包括在来自碳燃料的废料中和在其他熔融介质例如氢氧化钠和离子液体中存在的污染物的影响的一系列应用。
本发明提供了一种方法,其显示出观察和分析不同污染物对在直接碳燃料电池中碳的氧化反应的影响的能力。使用该方法以证明将煤污染物的包含到固体电极工作电极区域对石墨碳模型燃料的氧化机理具有显著的影响,并表明煤灰对直接碳燃料电池中的不同煤的期望性能的相互作用和重要性。
黏土材料表现为作为用于石墨碳的氧化的催化剂,而石英可以严重地抑制碳的氧化行为。此外,尽管添加石英会导致在电池内额外的电化学响应,但发现当向DCFC的碳酸盐电解质添加低浓度的特定灰分组分时,其对氧化反应没有不利影响。
尽管已经参考具体实施例描述了本发明,但是本领域技术人员会理解本发明可以以许多其他形式具体实现。例如,如示例的石墨电极代表本发明更通常涉及的碳电极的特别优选的形式。
在该文件和其权利要求中,元素前不使用数量词时,不排除多于一个元素存在的可能性,除非文中清楚地指出有且仅有一个元素。因此不使用数量词时通常指“至少一个”。
参考文献
[1]S.Giddey,S.P.S.Badwal,A.Kulkarni and C.Munnings,Progress in Energyand Combustion Science,2012,38,360-399.
[2]X.Li,Z.H.Zhu,R.De Marco,J.Bradley and A.Dicks,J.Phys.Chem.A,2010,114,3855-3862.
[3]X.Li,Z.H.Zhu,R.De Marco,J.Bradley and A.Dicks,Journal of Power Sources,2010,195,4051-4058.
[4]R.D.Weaver,N.T.I.Service and U.S.N.T.I.Service,Direct Electrochemical Generation of Electricity from Coal:Quarterly Progress Report,Stanford Research Institute,1979.
[5]A.C.Rady,S.Giddey,S.P.S.Badwal,B.P.Ladewig and S.Bhattacharya,Energy Fuels,2012,26,1471-1488.
[6]D.Cao,Y.Sun and G.Wang,Journal of Power Sources,2007,167,250-257.
[7]T.M.Gür,Chemical Reviews,2013,113,6179-6206.
[8]X.Y.Xu,W.Zhou,F.L.Liang and Z.H.Zhu,Int.J.Hydrog.Energy,2013,38,5367-5374.
[9]L.Guo,J.M.Calo,E.DiCocco and E.J.Bain,Energy Fuels,2013,27,1712-1719.
[10]A.Elleuch,J.Yu,A.Boussetta,K.Halouani and Y.Li,Int.J.Hydrog.Energy,2013,38,8514-8523.
[11]B.Cantero-Tubilla,C.C.Xu,J.W.Zondlo,K.Sabolsky and E.M.Sabolsky,Journal of Power Sources,2013,238,227-235.
[12]A.Kulkarni,S.Giddey and S.P.S.Badwal,Solid State Ion.,2011,194,46-52.
[13]Y.Nabae,K.D.Pointon and J.T.S.Irvine,Journal of the Electrochemical Society,2009,156,B716-B720.
[14]G.A.Hackett,J.W.Zondlo and R.Svensson,Journal of Power Sources,2007,168,111-118.
[15]Y.Nabae,K.D.Pointon and J.T.S.Irvine,Energy Environ.Sci.,2008,1,148-155.
[16]C.Q.Wang,J.Liu,J.Zeng,J.L.Yin,G.L.Wang and D.X.Cao,Journal of Power Sources,2013,233,244-251.
[17]N.J.Cherepy,R.Krueger,K.J.Fiet,A.F.Jankowski and J.F.Cooper,Journal of the Electrochemical Society,2005,152,A80-A87.
[18]X.Li,Z.H.Zhu,R.De Marco,A.Dicks,J.Bradley,S.M.Liu and G.Q.Lu,Ind.Eng.Chem.Res.,2008,47,9670-9677.
[19]D.G.Vutetakis,D.R.Skidmore and H.J.Byker,Journal of the Electrochemical Society,1987,134,3027-3035.
[20]S.S.J.Warne,Thermochimica Acta,1996,272,1-9.
[21]K.E.Benfell,B.B.Beamish and K.A.Rodgers,Thermochimica Acta,1996,286,67-74.
[22]F.J.Maldonado-Hódar,J.Rivera-Utrilla,A.M.Mastral-Lamarca and M.A.Ferro-García,Fuel,1995,74,818-822.
[23]F.Moodi,A.A.Ramezanianpour and A.S.Safavizadeh,Scientia Iranica,2011,18,906-912.
[24]G.J.Janz and M.R.Lorenz,Journal of Chemical and Engineering Data,1961,6,321-323.
[25]L.Lu,V.Sahajwalla,C.Kong and D.Harris,Carbon,2001,39,1821-1833.
[26]M.Castellano,A.Turturro,P.Riani,T.Montanari,E.Finocchio,G.Ramis and G.Busca,Applied Clay Science,2010,48,446-454.
[27]C.R.Ward,International Journal of Coal Geology,2002,50,135-168.
[28]T.Koishi,S.Kawase,S.Tamaki and T.Ebisuzaki,Journal of the Physical Society of Japan,2000,69,3291-3296.
[29]J.P.Hansen and I.R.McDonald,Journal of Physics C-Solid State Physics,1974,7,L384-L386.
[30]G.J.Janz,Molten Salts Handbook,Academic Press,New York,1967.
[31]P.L.Spedding and R.Mills,Journal of the Electrochemical Society,1965,112,594-&.
[32]M.F.Costa,Journal of Molecular Liquids,2008,138,61-68.
[33]M.Chen,C.Wang,X.Niu,S.Zhao,J.Tang and B.Zhu,Int.J.Hydrog.Energy,2010,35,2732-2736.
[34]S.V.Devyatkin and A.D.Pisanenko,Russian Journal of Applied Chemistry,2002,75,562-564.
- 还没有人留言评论。精彩留言会获得点赞!