电极催化剂的制造方法以及电极催化剂与流程
本发明涉及制造适宜用于燃料电池的电极催化剂的方法。另外,本发明涉及电极催化剂。
背景技术:
固体高分子型燃料电池具备膜电极接合体,该膜电极接合体以全氟烷基磺酸型高分子等具有质子传导性的高分子膜作为固体电解质,在该固体高分子膜的各面上形成了施加了电极催化剂而成的氧极以及燃料极。
电极催化剂通常是在作为载体的炭黑等导电性碳材料的表面上担载以铂为主的各种贵金属催化剂而成。已知电极催化剂根据燃料电池的运转时的电位变化,碳发生氧化腐蚀,引起所担载的金属催化剂的凝聚或脱落。其结果是,随着运转时间的经过,燃料电池的性能不断降低。因此,在燃料电池的制造中,通过使多于实际上所需的量的贵金属催化剂担载在载体上,性能方面具有富裕,实现高寿命化。但是,从经济性的观点出发,不能说这是有利的。
因此,为了实现固体高分子型燃料电池的高性能化、高寿命化或经济性的改善等,进行了关于电极催化剂的各种研讨。例如,提出了使用作为非碳系材料的导电性氧化物载体来代替一直以来作为载体使用的导电性碳(参考专利文献1)。该文献中,作为电极催化剂的载体,使用氧化锡。在该载体的表面上担载有铂等贵金属的微粒。该文献中记载了该电极催化剂具有优良的电化学的催化活性,并且具有高耐久性。该文献中,贵金属的微粒是通过将贵金属的胶体在还原气氛下、在80℃至250℃下进行热处理而生成。
现有技术文献
专利文献
专利文献1:us2010/233574a1
技术实现要素:
关于专利文献1中记载的电极催化剂的性能,本发明人进行了研讨,结果发现,活性支配电流密度等催化性能存在改善的余地。因此,认为通过使催化活性高于铂的铂镍合金担载在氧化锡上,电极催化剂的催化性能会提高,通过专利文献1中记载的方法,尝试制造在氧化锡的表面上担载了铂镍合金而成的电极催化剂。但是,在该文献记载的方法中,为了使镍固溶在铂中,需要将铂金属以及镍金属的前体担载在氧化锡上而成的试样在还原气氛下、在例如200℃以上的高温下进行热处理,因此在热处理时铂不仅与镍发生合金化、而且与氧化锡中的锡发生合金化,没有得到所期望的催化性能。
因此,本发明的课题在于,提供能够制造能消除上述以往技术所具有的各种缺点的电极催化剂的方法。
本发明提供一种电极催化剂的制造方法,其具备:
分散液制作工序,该工序是将(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂、(ii)金属氧化物的催化剂载体粉、(iii)铂化合物和(iv)过渡金属化合物混合,制作分散液;
担载工序,该工序是加热上述分散液,使铂和过渡金属的铂合金担载在上述催化剂载体粉的表面上;
固液分离工序,该工序是从上述担载工序后的上述分散液中分离分散相(分散质),得到在上述催化剂载体粉上担载了上述铂合金而成的催化剂粉;和
加热处理工序,该工序是将上述催化剂粉在真空下或在还原气体气氛下进行加热。
另外,本发明提供一种电极催化剂,其是在氧化锡的载体上担载了铂和过渡金属的铂合金粒子而成的电极催化剂,
上述铂合金粒子包含表面区域和与该表面区域相比位于中心侧的中心区域,在上述表面区域中具有铂富集层,
将上述电极催化剂进行x射线衍射测定而得到的衍射图形中,铂合金的(200)面的峰的衍射角2θ为46.5°~48.0°,
通过x射线光电子分光分析法测定的上述电极催化剂的表面及其附近的分析区域中的由下述式(1)定义的sn的金属化率为5%以下。
sn的金属化率(%)=rsn-金属/(rsn-金属+rsn-氧化物)×100(1)
rsn-金属:通过x射线光电子分光分析法测定的来自sn3d5/2轨道的光谱中sn金属所占的面积,
rsn-氧化物:通过x射线光电子分光分析法测定的来自sn3d5/2轨道的光谱中sn氧化物所占的面积。
附图说明
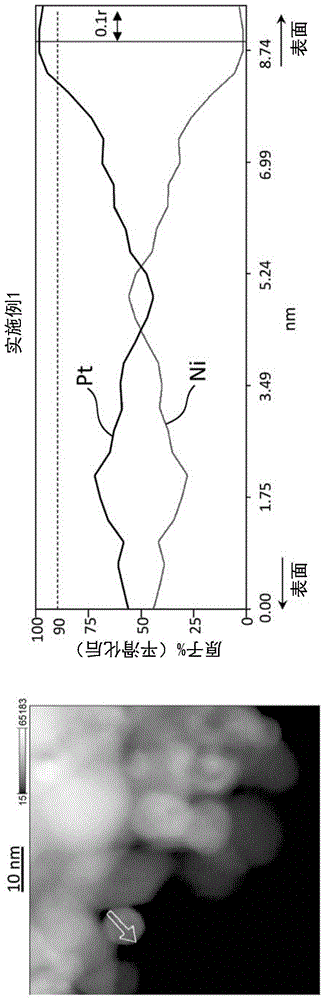
图1是使用了以实施例1中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图2是使用了以实施例2中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图3是使用了以实施例3中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图4是使用了以实施例5中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图5是使用了以实施例7中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图6是使用了以实施例8中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图7是使用了以比较例1中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图8是使用了以比较例2中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图9是使用了以比较例3中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图10是使用了以比较例4中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图11是使用了以比较例5中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图12是使用了以比较例6中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
图13是使用了以比较例7中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。
具体实施方式
以下,基于其优选的实施方式对本发明进行说明。本发明中作为制造对象的电极催化剂具有在载体的表面上担载了催化剂的结构。作为载体,适合使用具有导电性的金属氧化物。本发明中“具有导电性”是指金属氧化物在57mpa的压力下的体积电阻率为1×104ω·cm以下。作为在载体的表面上担载的催化剂,适合使用铂和过渡金属的铂合金。通过本发明的方法制造的电极催化剂适合作为各种燃料电池的催化剂使用。作为这样的燃料电池,可以列举例如固体高分子型燃料电池作为典型的例子。
本发明的制造方法具有:分散液制作工序,该工序是将(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂、(ii)金属氧化物的催化剂载体粉、(iii)铂化合物和(iv)过渡金属化合物混合,制作分散液;担载工序,该工序是加热上述分散液,使铂和过渡金属的铂合金担载在上述催化剂载体粉的表面上;固液分离工序,该工序是从上述担载工序后的上述分散液中分离分散相,得到在上述催化剂载体粉上担载了上述铂合金而成的催化剂粉;和加热处理工序,该工序是将上述催化剂粉在真空下或在还原气体气氛下进行加热。
关于通过使用本发明的制造方法能够得到催化性能优良的电极催化剂的理由,本发明人认为是由于,通过加热分散液,利用上述溶剂的还原作用,在更低温下还原铂化合物以及过渡金属化合物,能够生成两者的合金,从而能够抑制铂与构成催化剂载体的金属氧化物的该金属元素的合金化。另外,本发明人认为是由于,通过将固液分离工序后的催化剂粉在真空下或在还原气体气氛下加热,铂和过渡金属的铂合金粒子的表面的合金状态发生变化,形成催化活性更高的铂合金,由此催化性能进一步提高。
本发明的制造方法大致分为(1)分散液制作工序、(2)担载工序、(3)固液分离工序和(4)加热处理工序。以下对各种工序进行详细说明。
(1)分散液制作工序中,分散液是通过将以下(i)~(iv)作为其构成成分混合来制作。
(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂。
(ii)金属氧化物的催化剂载体粉。
(iii)铂化合物。
(iv)过渡金属化合物。
关于(i)~(iv),例如能够将它们一起投入容器等中,进行混合。或者,可以在(i)中添加(ii)~(iv)来进行混合。添加的顺序在本发明中不是临界的,可以根据各成分的性状或配合比率来适当确定添加的顺序。
在分散液制作工序中,优选以相对于作为目标的分散液的总质量使(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂的比例优选为30质量%~99.9质量%、进一步优选为40质量%~99.7质量%、更优选为50质量%~99质量%的方式添加该溶剂。需要说明的是,在发挥本发明的效果的限度内,在分散液中可以含有上述溶剂以外的溶剂。
在分散液制作工序中,优选以相对于作为目标的分散液的溶剂体积使(ii)金属氧化物的催化剂载体粉的比例优选为0.1g/l~500g/l、进一步优选为1g/l~150g/l、更优选为2g/l~100g/l的方式添加该载体粉。
在分散液制作工序中,优选以相对于作为目标的分散液的溶剂体积使(iii)铂化合物的比例优选为2.5×10-4mol/l~1.2mol/l、进一步优选为6.0×10-4mol/l~8.0×10-1mol/l、更优选为1.5×10-3mol/l~8.0×10-2mol/l的方式添加该铂化合物。
在分散液制作工序中,优选以相对于作为目标的分散液的溶剂体积使(iv)过渡金属化合物的比例优选为4.0×10-4mol/l~2.0×10-1mol/l、进一步优选为6.0×10-4mol/l~1.2×10-1mol/l、更优选为2.0×10-3mol/l~6.0×10-2mol/l的方式添加该过渡金属化合物。
在分散液制作工序中,除了混合上述成分之外,还可以混合包含羧基的芳香族化合物。通过在分散液中含有包含羧基的芳香族化合物,从而在(2)担载工序中,更加顺利地进行铂以及过渡金属的还原,由此铂合金的固溶状态变得更加均匀。其结果是,作为目标的电极催化剂的催化性能进一步提高。
在分散液制作工序中,优选以相对于作为目标的分散液的溶剂体积使上述包含羧基的芳香族化合物的比例优选为4.0×10-4mol/l~4.0mol/l、进一步优选为2.0×10-2mol/l~3.0mol/l、更优选为4.0×10-2mol/l~2.0mol/l的方式添加该芳香族化合物。
在分散液制作工序中,(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂被使用作为将(iii)铂化合物或(iv)过渡金属化合物溶解的溶剂。另外,也被使用作为将(iii)铂化合物或(iv)过渡金属化合物还原的还原剂。从这些观点出发,作为(i)的亚砜化合物的溶剂,可以列举出例如二甲基亚砜等。作为酰胺化合物的溶剂,可以列举出例如:n-甲基-2-吡咯烷酮、n-乙基-2-吡咯烷酮、n-丙基-2-吡咯烷酮以及n-羟乙基-2-吡咯烷酮等内酰胺化合物(分子内环状酰胺化合物)、以及n,n-二甲基甲酰胺、n-甲基甲酰胺、n,n-二乙基甲酰胺、n-乙基甲酰胺、n,n-二甲基乙酰胺以及n,n-二乙基乙酰胺等非环状酰胺化合物。这些溶剂中,为了容易形成活性支配电流密度等催化性能优良的电极催化剂,优选为作为酰胺化合物一种的具有甲酰胺基的有机化合物的溶剂、即甲酰胺、n,n-二甲基甲酰胺、n-甲基甲酰胺、n,n-二乙基甲酰胺、n-乙基甲酰胺等。这些溶剂可以单独使用一种,或者可以组合使用两种以上。
在分散液制作工序中,(ii)金属氧化物的催化剂载体粉由金属氧化物的载体的粒子(以下也称为“载体粒子”)的集合体形成。作为载体粒子,可以使用具有导电性的金属氧化物的粒子。作为具有导电性的金属氧化物,可以列举出例如:铟系氧化物、锡系氧化物、钛系氧化物、锆系氧化物、硒系氧化物、钨系氧化物、锌系氧化物、钒系氧化物、钽系氧化物、铌系氧化物以及铼系氧化物。作为进一步优选的无机氧化物,可以列举出例如:在锡氧化物中包含氟以及氯等卤素、铌、钽、锑、钨中的一种以上元素的化合物。具体而言,可以列举出:含锡的铟氧化物、含锑的锡氧化物、含氟的锡氧化物、含钽的锡氧化物、含锑以及钽的锡氧化物、含钨的锡氧化物、含氟以及钨的锡氧化物以及含铌的锡氧化物这样的含(掺杂)金属或非金属的锡氧化物等。但从固体高分子型燃料电池的发电环境下的物质的稳定性的观点出发,特别优选载体粒子为包含氧化锡的陶瓷材料。
由具有导电性的金属氧化物形成的载体粒子可以通过各种方法制造。制造方法大致分为湿式法以及干式法。从制造微粒的载体粒子的观点出发,采用湿式法是有利的。作为湿式法的一个例子,为了制造由含有卤素的氧化锡形成的载体粒子,优选采用以下的方法。该制造方法的详细情况例如在wo2016/098399中所记载。
载体粒子的一次粒径优选为5nm~200nm,进一步优选为5nm~100nm,更优选为5nm~50nm。载体的一次粒径可以通过由电子显微镜图像或小角x射线散射测定的载体的一次粒径的平均值得到。例如,用电子显微镜图像观察载体粒子,以500个以上的粒子作为对象,测定最大横断长,算出其平均值,由此求得载体的一次粒径。
在分散液制作工序中,作为(iii)铂化合物,优选使用在(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂中能够溶解的铂化合物,但不限于此。作为(iii)铂化合物,可以使用例如铂络合物或铂盐。作为铂化合物的具体例,可以列举出:作为铂络合物之一的双(乙酰基丙酮)铂(ii)、六氯化物铂(iv)酸、四氯化物铂(ii)酸、二硝基二氨合铂(ii)、二氯四氨合铂(ii)水合物、六羟铂(iv)酸等。
在分散液制作工序中,作为(iv)过渡金属化合物,优选使用在(i)选自亚砜化合物以及酰胺化合物中的至少一种溶剂中能够溶解的化合物,但不限于此。作为(iv)过渡金属化合物,可以使用例如过渡金属络合物或过渡金属盐。作为过渡金属的例子,可以列举出镍、钴、铁、铬、钛、钒、锰、铜、锌、钪等,但不限于这些。另外,过渡金属化合物可以单独使用一种,或者也可以组合使用两种以上。以上的过渡金属之中,从与铂的合金的催化活性高的观点出发,优选使用镍、钴、铁或铬的化合物。
如后所述,(i)溶剂为具有甲酰胺基的有机化合物的溶剂的情况下,(iv)过渡金属化合物也可以是具有结晶水的化合物。作为不具有结晶水的过渡金属化合物,可以列举出例如:作为镍络合物之一的双(2,4-戊二酮酸)镍(ii),作为具有结晶水的过渡金属化合物,可以列举出例如:作为镍络合物之一的双(2,4-戊二酮酸)镍(ii)二水合物以及六氟乙酰基丙酮镍(ii)水合物、或者例如作为镍盐的乙酸镍四水合物、硝酸镍六水合物以及氯化镍六水合物等。
在分散液制作工序中,附加使用的上述包含羧基的芳香族化合物是具有至少一个芳香族环、并且具有至少一个与芳香族环直接键合或通过键合基团间接地键合的羧基的化合物。作为芳香族环,可以列举出例如苯环、萘环以及蒽环等。另外,包含至少一个氮或氧并且具有芳香族性的杂环也包含在芳香族环的范畴中。作为包含羧基的芳香族化合物的具体例子,可以列举出苯甲酸、邻苯二甲酸、对苯二甲酸、水杨酸、乙酰基水杨酸等。这些芳香族化合物可以单独使用一种,或者也可以组合使用两种以上。
通过在分散液中包含附加使用的上述包含羧基的芳香族化合物,从而在(2)担载工序中,更加顺利地进行铂以及过渡金属的还原,由此,铂合金的固溶状态变得更加均匀。其结果是,作为目标的电极催化剂的催化性能进一步提高。
将使用以上的各成分来制备的分散液用于(2)担载工序,使由铂和过渡金属的铂合金形成的催化剂担载在催化剂载体粉的粒子的表面上。催化剂的担载是通过加热分散液来实现。分散液的加热可以在大气压、开放下进行,或者也可以在密闭下进行。在大气压、开放下进行加热的情况下,可以在使挥发成分回流的同时进行加热。
从能够使催化剂在载体粒子的表面上均匀地担载的观点出发,优选的是在担载工序中的分散液的加热前,使在分散液中包含的各成分充分均匀地分散。为了该目的,优选的是在担载工序中的分散液的加热前,进行对分散液施加超声波的分散处理。利用超声波的分散处理优选在非加热下进行,例如优选在15℃~25℃下进行。
在上述分散液的分散处理后,并且担载工序中的分散液的加热前,优选对分散液进行预搅拌。预搅拌主要是为了载体与其他原料的均匀混合和前体向载体上吸附的目的而进行的。例如,预先将具有磁性的搅拌子沉入分散液中,通过在外部设置的磁性搅拌装置使其旋转,由此可以进行预搅拌。或者,也可以通过在分散液中配置搅拌翼并使其旋转来进行预搅拌。在上述的任一种情况下,预搅拌均优选在非加热下进行,例如优选在15℃~25℃以下进行。
预搅拌的时间根据分散液的体积、粘度等性状、分散液中包含的催化剂载体粉的浓度等适当设定,通常优选为30分钟~120小时,进一步优选为4小时~120小时,更优选为12小时~120小时。
预搅拌可以在各种气氛下进行。例如,可以在大气下等含氧气氛下、氩气或氮气等不活泼气体气氛下进行。从不会使还原铂化合物以及过渡金属化合物的还原剂失活的观点出发,优选在不活泼气体气氛下对分散液进行预搅拌。
预搅拌结束后,转移至担载工序,加热分散液。与预搅拌同样地,分散液的加热也可以在各种气氛下进行。例如可以在大气下等含氧气氛下、氩气或氮气等不活泼气体气氛下进行。从不会使还原铂化合物以及过渡金属化合物的还原剂失活的观点、以及不会使在载体粒子上担载的催化剂的活性尽可能降低的观点出发,优选在不活泼气体气氛下加热分散液。从操作的简便化的观点出发,优选预搅拌的气氛与加热的气氛相同。
在担载工序中,分散液的加热温度可以根据分散液中包含的各成分的种类和配合比率适当选择,通常优选为大于等于120℃且小于200℃,进一步优选为120℃~175℃。以该加热温度为条件,加热时间优选为3小时~120小时,进一步优选为6小时~72小时,更优选为12小时~48小时。
通过加热分散液,在分散液中包含的铂化合物以及过渡金属化合物发生热分解,与此同时铂以及过渡金属被还原,生成两者的合金。生成的铂合金附着在载体粒子的表面上,得到作为目标的电极催化剂。
在担载工序中,以作为催化剂的铂和过渡金属的铂合金的担载量相对于电极催化剂的质量优选为0.1质量%~50质量%、进一步优选为1质量%~30质量%的方式来担载催化剂。催化剂的担载量的调节通过下述方式来实现:适当调节分散液制作工序中金属氧化物的催化剂载体粉、铂化合物以及过渡金属化合物的浓度,与此同时在担载工序中控制例如加热温度和加热时间。
从进一步提高这样得到的电极催化剂的催化性能、特别是活性支配电流密度的观点出发,本发明人经过研讨,结果发现,在担载工序中,在分散液中存在甲酸化学种的状态下加热分散液是有利的。通过在甲酸存在下加热分散液来使电极催化剂的催化性能进一步提高的理由目前尚不明确,本发明人认为也许是由于,通过甲酸化学种促进在分散液中包含的铂化合物以及过渡金属化合物的还原,由此铂合金的固溶状态变得更加均匀。
作为甲酸化学种,可以列举出例如:甲酸(hcooh)、甲酸盐((hcoo)nx(x表示阴离子,n表示x的价数))、甲酸根离子(hcoo-)等。这些甲酸化学种可以分别单独使用,或者也可以组合使用两种以上。关于在加热中的分散液中存在的甲酸化学种的总浓度,相对于分散液的溶剂体积,优选为1×10-5mol/l~1mol/l,进一步优选为1×10-4mol/l~1×10-1mol/l,更优选为1×10-3mol/l~1×10-1mol/l。通过甲酸化学种以该浓度范围存在于分散液中,作为目标的电极催化剂中的活性支配电流密度等催化性能进一步提高。
加热中的分散液中存在的甲酸化学种的总浓度例如可以通过液相色谱等来测定。
为了使甲酸化学种存在于加热中的分散液中,作为一个例子可以列举出(a)在分散液中添加规定量的甲酸化学种的方法。作为其他方法,可以列举出:(b)方法,其是在分散液制作工序中,作为溶剂使用具有甲酰胺基的有机化合物的溶剂,与此同时混合水或含水化合物,使水存在于制作后的分散液中。根据(b)方法,通过在水或含水化合物存在于分散液中的状态下加热分散液,分散液中包含的溶剂即(i)包含具有甲酰胺基的有机化合物的溶剂与水发生反应,生成甲酸化学种(例如甲酸)。
如果采用上述(b)方法,则与采用(a)方法的情况相比,在担载工序中的加热过程中,分散液中的甲酸的存在量缓慢增加,因此铂与过渡金属的合金化容易缓慢地进行。由此,具有如下优点:能够制造铂原子与过渡金属原子更加均匀地进行合金化、与此同时铂合金的微粒均匀分散地担载在载体粒子的表面上而成的催化剂。
在采用上述(b)方法的情况下,作为含水化合物,可以使用通过分散液的制作而能够使水存在于分散液中的化合物。作为这样的化合物,例如作为在分散液中包含的物质即(iv)过渡金属化合物,通过使用具有结晶水的物质,该物质兼具过渡金属化合物和含水化合物,因此是简便的。通过使用具有结晶水的过渡金属化合物来制作分散液,在分散液中结晶水溶出,从而在分散液中存在水。该水在分散液的加热工序中与作为溶剂的(i)具有甲酰胺基的有机化合物反应。该反应的结果是,在分散液中生成甲酸化学种。
在采用上述(b)方法的情况下,从进一步促进铂与过渡金属的合金化、并且能够制造铂原子与过渡金属原子更加均匀地合金化而成的电极催化剂的观点出发,相比于添加水而言添加具有结晶水的过渡金属化合物是更有利的。其理由在于,添加具有结晶水的过渡金属化合物时,在过渡金属离子的周围水容易配位,由此在担载工序时产生的甲酸也容易配置在过渡金属离子的周围。由此,本发明人认为是由于,在比铂更难还原的元素即过渡金属离子的周围,还原剂更多地存在,因此铂与过渡金属的合金化被进一步促进。
在采用上述(b)方法的情况下,关于水或含水化合物的添加量,优选为分散液中存在的水的浓度相对于分散液的溶剂体积达到1×10-3mol/l~10mol/l的量,进一步优选为达到1×10-2mol/l~5mol/l的量,更优选为达到1×10-2mol/l~5×10-1mol/l的量。通过采用这样的添加量,能够使甲酸化学种在分散液中以期望的浓度存在。
在(2)担载工序中,在载体粒子的表面上担载催化剂后,就接着进行(3)固液分离工序。本工序中,从担载工序后的分散液中分离出作为分散相的电极催化剂。通过该工序,得到在催化剂载体粉上担载了铂和过渡金属的铂合金而成的催化剂粉即电极催化剂。固液分离工序中,可以没有特别限定地使用公知的各种固液分离方法。例如可以列举出使用过滤器进行的过滤、离心分离以及倾析等。
这样地操作而使得电极催化剂被分离后,就接着进行(4)加热处理工序。本制造方法中,优选的是在该加热处理工序前进行浸提,除去在铂合金粒子的表面上存在的微量的杂质。通过进行该操作,电极催化剂的催化活性进一步提高。浸提通常是通过对电极催化剂进行酸处理来实现。通过酸处理,在铂合金粒子的表面上存在的微量的杂质溶出而被除去。作为用于浸提的酸,例如可以列举出:高氯酸、硝酸、硫酸、盐酸以及过氧化氢等。酸的浓度优选为0.01mol/l~10mol/l,进一步优选为0.1mol/l~1mol/l。
浸提可以在加热分散液后的状态下进行,或者也可以在非加热下进行。在加热下进行浸提的情况下,可以将分散液优选在30℃~100℃以下加热。非加热下的浸提可以在例如15℃~25℃以下进行。关于浸提的时间,例如在加热下的情况下,可以设定为0.1小时~100小时。
浸提后的电极催化剂被用于(4)加热处理工序,催化性能进一步提高。加热处理可以在例如真空下或在还原气体气氛下进行。通过将电极催化剂用于加热处理,铂和过渡金属的铂合金粒子的表面的合金状态发生变化,铂合金的催化活性进一步提高。
这样一来,通过用于(4)加热处理工序,电极催化剂的催化性能提高的理由目前尚不明确,但本发明人认为也许是由于,通过加热处理工序使铂在铂合金粒子的表面上移动,由此形成铂富集了的层(铂富集层)。
在真空下对电极催化剂进行加热处理的情况下,体系内的真空的程度以绝对压计优选为10pa,进一步优选为1×10-3pa。在还原气体气氛下对电极催化剂进行加热处理的情况下,优选使还原气体在体系内流通的状态下加热。作为还原气体,可以使用例如氢气、一氧化碳气体以及它们的混合气体等。
关于将电极催化剂在真空下加热时的温度,从铂合金的催化活性更进一步提高的观点出发,优选设定为上述(2)担载工序中的加热温度以上。特别是本工序中的加热温度优选设定为60℃~400℃,进一步优选设定为60℃~200℃,其中,更优选为(2)担载工序中的加热温度以上。将电极催化剂在还原气体气氛下加热时的温度优选设定为小于200℃,进一步优选设定为60℃以上且小于200℃。在上述的任一种情况下加热时间均优选设定为0.1小时~20小时,进一步优选设定为0.5小时~10小时。
通过如上所述的方法,能够容易地制造活性支配电流密度等催化性能优良的电极催化剂。
在作为载体例如使用氧化锡的情况下,通过如上所述的方法得到的电极催化剂(以下,称为本发明的电极催化剂)具有在氧化锡的载体上担载了铂和过渡金属的铂合金粒子而成的构成。作为电极催化剂的性状,可以列举出例如粉末状。
本发明的电极催化剂中的铂合金粒子包含表面区域和与该表面区域相比位于中心侧的中心区域,如上所述,表面区域中的铂的存在比例大于中心区域中的铂的存在比例。
表面区域是指铂合金粒子的表面及其附近的区域,在将铂合金粒子视为球的情况下,将其半径设定为r时,是指距离球的中心为0.8r~r的区域。中心区域是指铂合金粒子的中心及其附近的区域,在将铂合金粒子视为球的情况下,将其半径设定为r时,是指距离球的中心为0~0.3r的区域。
“铂合金粒子的表面区域中的铂的存在比例大于中心区域中的铂的存在比例”是指表面区域中的铂原子比例的平均值高于中心区域中的铂原子比例的平均值。
铂的存在比例是指铂的原子数相对于铂与过渡金属的合计原子数的比例、即“铂的存在比例=铂的原子数/(铂的原子数+过渡金属的原子数)×100”,其单位为原子%。
该铂的存在比例是通过利用扫描透射型电子显微镜附带的能量分散型x射线分光法(stem-eds)的线性分析求得。在该线性分析中,表面区域中的铂原子比例的平均值是通过将在距离中心为0.8r的位置与距离中心为r的位置之间存在的符合stem-eds的分辨能力的全部测定点的铂原子比例进行平均而得到的。在假设分析对象线横穿粒子的情况下,表面区域在该线的两端侧存在两个部位。在该情况下,关于其两端侧的两个部位,分别按上述一定距离求出铂的比例,得到全部的比例的平均值。另外,中心区域中的铂原子比例的平均值是将距离中心为0的位置与距离中心为0.3×r的位置之间存在的符合stem-eds的分辨能力的全部测定点的铂原子比例进行平均而得到的。“表面区域的铂比例大于中心区域的铂比例”通过沿着电极催化剂中的任意的铂合金粒子之一的至少一个线的线性分析显示即可,在该情况下,就假设沿着其他线的线性分析而言,即使表面区域的铂比例为中心区域的铂比例以下,也显示出表面区域的铂比例大于中心区域的铂比例。
另外,本发明的电极催化剂中的铂合金粒子在其表面区域具有铂富集层。铂富集层是指在将铂合金粒子视为球、将其半径设定为r的情况下,铂的存在比例为90原子%以上的区域连续存在0.1r以上的区域。铂富集层可以为例如大致球壳的形状。或者,铂富集层可以不连续地存在于表面区域上。从提高电极催化剂的催化性能的观点出发,铂富集层优选以大致球壳的形状存在于表面区域。
在表面区域是否具有铂富集层可以通过利用stem-eds的线性分析按照如下所示地进行确认。
首先,以铂合金粒子为对象,进行利用stem-eds的映射测定后,从所得的eds映射数据中,抽取通过铂合金粒子的大致中心的任意线的轮廓数据进行分析,由此进行利用stem-eds的线性分析。接着,在将铂合金粒子视为以线性分析中的分析长度为直径的球,将其半径设定为r的情况下,沿着线性分析方向的表面区域(距离球的中心为0.8r~r的区域)中,通过确认铂的存在比例为90原子%以上的区域有无连续存在0.1r以上的区域,由此进行铂富集层的确认。
另外,本发明的电极催化剂中,通过x射线光电子分光分析法(xps)测定的电极催化剂的表面及其附近的分析区域中的由下述式(1)定义的sn的金属化率为5%以下、优选为3%以下。
sn的金属化率(%)=rsn-金属/(rsn-金属+rsn-氧化物)×100(1)
式中,rsn-金属表示通过x射线光电子分光分析法测定得到的来自sn3d5/2轨道的光谱中sn金属所占的面积。
rsn-氧化物表示通过x射线光电子分光分析法测定得到的来自sn3d5/2轨道的光谱中sn氧化物所占的面积。
此处所谓的表面及其附近的分析区域是指将电极催化剂用于利用x射线光电子分光分析法进行的测定时作为分析对象的深度方向的区域。利用xps进行的深度方向的测定距离、即表面及其附近的分析区域通常为从表面开始朝向深度方向的0nm~5nm的区域。锡的金属化率(%)具体而言是以在sn3d5/2的整个光谱的面积之中的来自sn金属的光谱的面积所占的比例来求得。
本发明中,xps优选在以下的(a1)~(a5)的条件下测定。(a1)x射线源:al的kα(hν=1486.6ev)
(a2)试样与检测器的角度:θ=45°
(a3)检测器的校正:使用cu2p和au4f来实施
(a4)分析区域:直径为0.1mm的圆
(a5)分析时的室压力:10-7~10-6pa等级
另外,本发明的电极催化剂中,在将该电极催化剂进行x射线衍射测定而得到的衍射图形中,铂合金的(200)面的峰的衍射角2θ为46.5°~48.0°。该电极催化剂反映出在载体上担载铂合金(pt-m(m表示过渡金属))粒子,在将该电极催化剂进行x射线衍射测定而得到的衍射图形中,铂合金的(200)面的峰的衍射角2θ从铂单质的46.2°向高角度侧移动。具体而言,上述衍射图形中的上述铂合金的峰的衍射角2θ为46.5°以上,由此,能够得到利用合金化实现的催化活性的提高效果。另外,上述铂合金的峰的衍射角2θ为48.0°以下。在比48.0°高的角度侧过渡金属向pt的固溶过度地进行(过渡金属为ni的情况下,铂合金中ni所占的摩尔浓度超过50%),催化活性降低。因此,通过设定为48.0°以下,具有能够保持高催化活性的优点。从这些方面出发,上述铂合金的峰的2θ为46.5°~48.0°,更优选为46.8°~47.7°。为了将上述铂合金的峰的2θ设定为上述值,在上述电极催化剂的制造方法中,在进行担载工序中的加热温度的控制的同时调节铂化合物以及过渡金属化合物的添加量即可。将峰的移动的程度以(200)面作为对象进行评价的理由是由于,可以避免与由作为载体的氧化锡所产生的衍射峰重合,容易进行分析。
从提高催化活性的观点出发,优选本发明的电极催化剂中的铂(pt)与过渡金属(m)的摩尔比pt/m为1以上。从提高铂的合金化率从而提高催化活性的观点出发,优选该摩尔比pt/m为10以下。从这些观点出发,摩尔比pt/m优选为1~10,更优选为1~7。从提高催化活性的观点出发,作为催化剂的铂和过渡金属的铂合金的合计担载量,相对于电极催化剂的质量,优选以达到0.1质量%~50质量%,进一步优选达到1质量%~30质量%的方式担载催化剂。催化剂的担载量的调节通过下述方式来实现:在上述制造方法中的分散液制作工序中适当调节金属氧化物的催化剂载体粉、铂化合物、过渡金属化合物的浓度,与此同时在担载工序中控制例如加热温度和加热时间。将电极催化剂通过适当的方法溶解,形成溶液,利用icp质谱对该溶液进行分析,由此可以求得铂以及过渡金属的担载量,铂和过渡金属的铂合金的担载量可以作为铂的担载量以及过渡金属的担载量的合计值来求得。
铂合金粒子以微粒的形式担载在载体的表面上是有利的。铂合金粒子的粒径例如优选为1nm~20nm,更优选为1nm~10nm,进一步优选为1nm~5nm。通过担载具有该范围的粒径的铂合金粒子,具有下述优点:能够有效地防止电极反应进行过程中铂合金粒子的溶出、另外也能够有效地防止铂合金粒子的比表面积的降低。铂合金粒子的粒径可以通过由电子显微镜图像或小角x射线散射测定的铂合金的粒径的平均值来得到。
铂合金粒子根据其担载量可以被覆载体的表面整个区域,也可以载体的表面的一部分露出的方式被覆。例如在以表面的一部分露出的方式被覆的情况下,可以仅被覆表面的一个部位,但在氧还原反应中相对于氧扩散量而言合金催化剂的反应面积如果过多,则导致氧扩散律速,可成为无法充分地发挥本来的催化活性的原因,因此优选保持适当的距离、以载体的表面露出的方式不连续地被覆。
本发明的制造方法中得到的电极催化剂可以在例如具有在固体高分子电解质膜的一个面上配置的氧极以及在另一个面上配置的燃料极的膜电极接合体中的氧极或燃料极中的至少一种中含有而使用。电极催化剂也可以适宜地在氧极以及燃料极这二者中含有。
特别是氧极以及燃料极优选包含:含有通过本发明的制造方法得到的电极催化剂的催化剂层;和气体扩散层。为了使电极反应顺利地进行,优选电极催化剂与固体高分子电解质膜相接。气体扩散层作为具有集流功能的支撑集流体发挥作用。另外,具有对电极催化剂充分地供给气体的功能。作为气体扩散层,可以使用与在该种技术领域中以往使用的气体扩散层同样的层。可以使用例如作为多孔材料的复写纸、碳布。具体而言,可以通过由将例如用聚四氟乙烯涂布表面后的碳纤维与没有进行该涂布的碳纤维设定为规定比例的线织成的碳布而形成。
作为固体高分子电解质,可以使用与在该种技术领域中以往使用的固体高分子电解质同样的物质。可以列举出例如:全氟磺酸聚合物系的质子传导体膜、在烃系高分子化合物中掺杂了磷酸等无机酸的膜、一部分用质子传导体的官能团取代后的有机/无机混合聚合物、在高分子基体中浸渗磷酸溶液或硫酸溶液后的质子传导体等。
上述膜电极接合体在其各面上配置隔膜而形成固体高分子型燃料电池。作为隔膜,可以使用例如在与气体扩散层的对向面上以隔开规定的间隔地形成向一个方向延伸的多个凸部(棱)的膜。相邻的凸部间形成断面为矩形的槽部。该槽部被使用作为燃料气体以及空气等氧化剂气体的供给排出用流路。燃料气体以及氧化剂气体分别从燃料气体供给单元以及氧化剂气体供给单元供给。在膜电极接合体的各面上配置的各个隔膜优选以在其上形成的槽部彼此垂直相交的方式配置。以上的构成形成了燃料电池的最小单位,可以由将该构成同时设置数十个~数百个而成的电池堆形成燃料电池。
以上,基于优选实施方式对本发明进行说明,但本发明不限于上述实施方式。例如,在上述实施方式中,以使用通过本发明的方法制造的电极催化剂作为固体高分子电解质型燃料电池的电极催化剂的例子为中心进行了说明,也可以使用通过本发明的方法制造的电极催化剂作为固体高分子电解质型燃料电池以外的燃料电池、例如碱型燃料电池、磷酸型燃料电池、直接甲醇型燃料电池等各种燃料电池中的电极催化剂。
实施例
以下,通过实施例对本发明更加详细地进行说明。但是,本发明的范围不限于该实施例。只要没有特别说明,则“%”是指“质量%”。
[实施例1]
(1)载体的制造工序
基于wo2016/098399中记载的实施例1,得到一次粒径为20nm的含钨/氟的氧化锡粒子。
(2)分散液制作工序
在容量为500ml的容量瓶中加入337ml的n,n-二甲基甲酰胺(简记:dmf、049-32363、和光纯药工业公司制)、9.87×10-3mol/l-dmf的双(乙酰基丙酮)铂(ii)(pt(acac)2、028-16853、和光纯药工业公司制)、7.40×10-3mol/l-dmf的双(2,4-戊二酮酸)镍(ii)(ni(acac)2、283657-25g、sigma-aldrich公司制)、2.49×10-1mol/l-dmf的苯甲酸(204-00985、和光纯药工业公司制),与此同时以10g/l-dmf的浓度加入(1)中所得到的载体。然后,在室温(25℃)下使用超声波分散器30分钟,使各成分混合而成的液体分散,得到分散液。需要说明的是,以“mol/l-dmf”或“g/l-dmf”作为单位的上述各值是指相对于作为分散液的溶剂的dmf的比例。
(3)担载工序
在使将加入了上述分散液的容量瓶用氩气吹扫后的状态得以保持的状态下,使其在室温的油中沉淀,将分散液以5℃/分钟的油升温速度升温到120℃。在将油浴中的油温控制为120℃的状态下,加热回流48小时。然后,移去油浴,冷却至室温后,进行过滤。接着,用丙酮与乙醇的混合溶剂(以体积比计为1:1的混合溶剂)清洗5次,用水与乙醇的混合溶剂(以体积比计为1:1的混合溶剂)清洗1次后进行干燥。使所得的干燥粉分散在400ml的0.5mol/l高氯酸中,在60℃下加热搅拌2小时,进行浸提。
(4)固液分离工序
将浸提后的分散液用过滤器过滤,接着清洗、干燥,得到担载了铂镍合金的电极催化剂。
(5)加热处理工序
使用连接有旋转泵的石英管状炉,将通过固液分离得到的干燥粉在180℃下经过2小时进行真空加热处理。由此得到担载了铂镍合金的电极催化剂。
将通过xps测定的来自sn3d5/2轨道的光谱中sn金属所占的浓度、通过icp质谱装置(icp-ms)测定的pt担载率(质量%)、ni担载率(质量%)、pt与ni的摩尔比[pt/ni]、通过粉末x射线衍射测定(xrd)得到的衍射图形中的铂合金的(200)面的峰的衍射角2θ(°)、利用使用了stem像以及eds映射结果的线性分析结果确认的铂富集层的有无、通过使用旋转盘电极(rde)的循环伏安法(cv:cyclicvoltammetry)以及对流伏安法(lsv:linearsweepvoltammetry)求出的0.64v(相对于ag/agcl)时的活性支配电流密度jk(ma/cm2-pt)的值示于以下的表1。需要说明的是,pt担载率以及ni担载率分别是用pt质量/(pt质量+ni质量+载体质量)×100、ni质量/(pt质量+ni质量+载体质量)×100定义的值。
xps测定通过下述方式来实施:使用ulvac-phi公司制versaprobeii,以x射线源为al的kα的单色光(hν=1486.6ev)、通能(passenergy)为55.0ev、能量阶跃为0.1ev、试样与检测器的角度为45°、试样的分析区域为直径为0.1mm的圆。分析软件使用multipak。
将来自sn3d5/2轨道的光谱中的主峰(锡氧化物的键合能量)的峰顶位置设定为486.7ev,进行带电校正。sn的金属化率通过将来自sn3d5/2轨道的光谱进行波形分离来求得。表示sn元素的金属键合状态的峰设定为键合能量超过484.5ev且小于485.2ev的峰,将其他作为表示sn的氧化物状态的峰来进行分离,计测它们的面积。使用这些,sn的金属化率根据上述式(1)由计算求得。
xrd是使用rigaku公司制rint-ttriii、使用cukα(0.15406nm、50kv、300ma)作为x射线源来进行测定。
利用stem-eds的线性分析如下进行。stem观察是使用日本电子公司制的扫描透射型电子显微镜(jem-arm200f),eds使用日本电子公司制的eds检测器(干燥sd100gv检测器×2台),分析软件使用thermofisherscientific公司制的nss4,在以下的测定条件下,实施stem观察与eds映射的测定。另外,线性分析由获得的eds映射数据抽取出通过铂合金粒子的大致中心的任意的线的轮廓数据,用上述分析软件来实施。
=测定条件=
·取样方法:将电极催化剂的粉末试样分散在mo栅格中
·加速电压:200kv(stem观察、eds均)
·观察倍率:300万倍
·探针尺寸:0.2nm
·映射像的像素数:256×256
·用于分析的元素的峰种:ptlα射线9.441kev
nikα射线7.771kev
·定量计算方法:cliff-lorimer法(薄膜近似法)
[实施例2]
除了将实施例1中的(2)分散液制作工序以及(3)担载工序中的dmf中的载体粒子的浓度、pt(acac)2的浓度、ni(acac)2的浓度以及苯甲酸的浓度、以及加热回流中的油温以及保持时间变更为表1所示的值以外,与实施例1同样操作,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表1。
[实施例3]
除了将实施例1中的(3)担载工序中的油温以及保持时间变更为表1所示的值以外,与实施例1同样操作,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表1。
[实施例4]
(1)载体的制造工序以及(5)加热处理工序与实施例1同样地进行。另外,将实施例1中的(2)分散液制作工序和(3)担载工序变更为以下的内容。
在容量为500ml的容量瓶中将337ml的n,n-二甲基甲酰胺(简记:dmf、049-32363、和光纯药工业公司制)、1.22×10-2mol/l-dmf的双(乙酰基丙酮)铂(ii)(pt(acac)2,028-16853、和光纯药工业公司制)、9.37×10-3mol/l-dmf的双(2,4-戊二酮酸)镍(ii)二水合物(ni(acac)2·2h2o、343-01981、同仁化学研究所公司制)、2.99×10-1mol/l-dmf的苯甲酸(204-00985、和光纯药工业公司制)以及(1)中得到的载体以16g/l-dmf的浓度加入后,在室温下使用30分钟超声波分散器,使其分散。接着,将装入了上述制作的分散液的容量瓶用氩气吹扫的同时,使用长径为约2cm、短径为约1cm的橄榄球型的搅拌子,在室温、400rpm下进行20小时的预搅拌。在将装入了分散液的容量瓶用氩气吹扫后的状态得以保持的状态下,使其在将油温控制为160℃的油浴中沉淀,开始升温。此时,容量瓶中的溶剂直至沸腾所需要的时间为约7分钟。沸腾后,在将油浴中的油温控制在160℃的状态下,加热回流12小时。然后,按照与实施例1同样的顺序,进行浸提、(4)固液分离工序以及(5)加热处理工序,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表1。
[实施例5至7]
除了将实施例4中的(5)加热处理工序中的加热温度变更为表1所示的值以外,与实施例4同样操作,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表1。
[实施例8]
将实施例3中的(5)加热处理工序的条件变更为氢气4体积%-氮气平衡气体气氛下的80℃、2小时的还原加热处理工序。除此以外,与实施例3同样操作,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表1。
[比较例1至4]
没有进行实施例1至4中的(5)加热处理工序。除此以外,与实施例1至4同样操作,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表2。
[比较例5]
将实施例4中的(5)加热处理工序的条件变更为氮气气氛下的180℃、2小时的热处理工序。除此以外,与实施例4同样操作,得到担载了铂镍合金的电极催化剂。将所得到的各物性示于表2。
[比较例6]
本比较例是基于专利文献1的记载、通过胶体法使铂镍合金催化剂担载的例子。
(1)载体的制造工序
与实施例1同样进行。
(2)担载工序
使5ml的h2ptcl6溶液(相当于pt1g)与蒸馏水295ml混合,通过15.3g的nahso3使铂还原后,用1400ml的蒸馏水进行稀释。接着,在加入naoh的5%水溶液来将ph调节至约5的同时,滴加35%过氧化氢(120ml),得到含有铂的胶体的液体。此时,适宜地添加naoh的5%水溶液,将液体的ph保持为约5。按照该顺序制备而得到的包含胶体的液体含有1g份的铂。在该液体中添加硝酸镍六水合物(ni(no3)2·6h2o)。添加量设定为1.49g以使得pt与ni的摩尔比即pt/ni达到1。然后,添加上述(1)中得到的载体8.7g,在90℃下混合3小时。混合后,冷却液体,进一步进行固液分离。为了从通过固液分离得到的含水粉体中除去氯化物离子,用1500ml的蒸馏水再次进行稀释,在90℃下进行1小时煮沸,冷却液体,进行固液分离。将该清洗作业实施4次。最后,在固液分离后,在大气下、在60℃下干燥12小时。由此,在载体的表面上担载了包含非定比氧化物的铂以及包含非定比氧化物的镍。将该载体在用氮气稀释后的4体积%氢气氛下、在200℃下进行2小时热处理,由此得到在载体上担载了铂镍合金而成的电极催化剂。将所得到的各物性示于表3。
[比较例7]
对于比较例6中得到的担载了铂镍合金的电极催化剂,进行实施例1中进行的(5)加热处理工序。将所得到的各物性示于表3。
[评价]
关于实施例、比较例中得到的电极催化剂,进行了使用旋转盘电极的循环伏安法(cv:cyclicvoltammetry)以及对流伏安法(lsv:linearsweepvoltammetry)。具体而言,按照以下的“电极制作”、“cv测定”以及“orr活性评价”的顺序进行操作。
电极制作
使用0.05μm的氧化铝糊,对直径为5mm的玻璃碳(gc)盘电极进行研磨,然后,使用纯水,进行超声波清洗。将担载了铂镍合金的试样加入90体积%乙醇水溶液中,用超声波匀质机使其分散。将其向gc盘上以该盘的单位面积的pt量达到12μg-pt/cm2-gc的密度进行涂布,在常温下使其干燥。干燥后,在gc盘上的催化剂上滴加5%nafion(注册商标)溶液(274704-100ml、sigma-aldrich公司制),以使膜厚达到50nm,常温下使其干燥。
cv测定
测定使用北斗电工株式会社制的电化学测定系统hz-7000来实施。在0.1mol/l的hclo4水溶液中吹扫1小时以上n2后,参比电极使用银-氯化银电极(ag/agcl),在电位范围为-0.25~0.742v(相对于ag/agcl)、扫描速度为0.5v/秒下实施300次清洁。然后,在电位范围为-0.25~0.74v下实施cv测定,作为主测定。电化学的活性表面积(ecsa:electrochemicalsurfacearea)的分析使用在0.4v以下被观察到的氢气的吸附波来实施。
orr活性评价
在cv测定中使用的上述电解液(hclo4水溶液)中吹扫氧气1小时以上后,进行lsv。在温度为25℃、电位范围为-0.20~1.00v(相对于ag/agcl)、扫描速度为10mv/秒下转速为400rpm至2500rpm,获取共计6条件的数据。将所得到的结果使用koutecky-levich图进行分析,得到在0.64v(相对于ag/agcl)时的活性支配电流密度jk(ma/cm2)的值。
表1
*在通过xps测定的sn3sd5/2光谱中sn金属所占的浓度
**jk为0.64v(相对于ag/agcl)时的评价值
表2
*在通过xps测定的sn3sd5/2光谱中sn金属所占的浓度
**jk为0.64v(相对于ag/agcl)时的评价值
表3
*在通过xps测定的sn3sd5/2光谱中sn金属所占的浓度
**jk为0.64v(相对于ag/agcl)时的评价值
由表1以及表2所示的结果可知,作为铂镍合金催化剂的加热处理进行了180℃下的真空加热处理的实施例1至4,与没有进行该处理的比较例1至4比较,判定各自活性支配电流密度jk增高。另外,由实施例4至7所示的结果判定,通过真空加热处理实现的效果不限于180℃的热处理温度。另外,由实施例8的结果判定,加热处理不仅可以在真空下,而且也可以在还原性气氛下进行。另一方面,由比较例5所示的结果判定,即使在氮气气氛下进行加热处理,也不能得到活性支配电流密度jk增高的效果。
另外,由表3所示的结果可知,比较例7的活性支配电流密度jk与比较例6比较,不会那么增高。由此判定:本发明的加热处理工序针对经由本发明的担载工序而制作的催化剂粉是有效的。
由表1以及表3所示的结果可知,实施例1至8与通过以往的胶体法制作的比较例6以及7比较,活性支配电流密度jk增高。
这样一来,作为通过在真空下或在还原性气氛下的铂镍合金催化剂的加热处理工序、该催化剂的活性支配电流密度jk增高的理由之一,本发明人推测是由于在铂镍合金的表面区域形成铂富集层。图1至6以及图7至13中示出使用以实施例1、2、3、5、7及8以及比较例1至7中得到的电极催化剂中的催化剂微粒作为对象的stem像以及eds映射结果而得到的线性分析结果。需要说明的是,图1至13的线性分析结果是将各自stem像中记载的从箭头的末端至顶端进行线性分析的结果,该箭头的末端的分析结果与分析图的左端对应。图7所示的比较例1中,仅仅在最表面铂的存在比例达到90原子%,但在将铂镍合金粒子视为球、将其半径设定为r的情况下,在表面区域中不存在至少0.1×r以上的区域中铂的存在比例为90原子%以上的铂富集层。关于比较例2~5,在表面区域也不存在铂富集层。相对于此,图1至6所示的实施例1、2、3、5、7以及8中,将铂镍合金粒子视为球、将其半径设定为r的情况下,在表面区域中至少0.1×r以上的区域观察到铂的存在比例为90原子%以上的铂富集层。关于作为其他实施例的实施例4以及6,也与实施例1同样地在铂镍合金的表面区域观察到铂富集层。另外,通过胶体法制作的担载了铂镍合金的电极催化剂即表3所示的比较例6以及比较例7中,即使进行与实施例1同样的加热处理,其活性支配电流密度jk也没有提高,如图12以及13所示,与其他比较例同样地在铂镍合金的表面区域没有观察到铂富集层的形成。本发明人推测,如果加热处理工序前的sn的金属化率高、即铂镍合金中的锡元素的比例高,则铂的富集过程会被阻碍,其结果是,无法得到利用加热处理带来的活性提高。
产业上的可利用性
根据本发明,能够容易地制造活性支配电流密度等的催化性能优良的电极催化剂。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于与催化剂结构一起使用的密封件的制作方法
- 用于烯烃二聚和/或低聚化的非负载型杂多酸金属盐催化剂的制作方法
- 一种双亲性含钴夹心型杂多酸及其应用
- 基于改性杂多酸催化剂的汽油烷基化脱硫方法及其产品的制作方法
- (S)-1-(2-羟基-1-苯乙基)硫脲修饰的Mn-Anderson型杂多酸催化剂、制备方法及其应用
- 稀土改性杂多酸催化剂的制备方法及生物柴油的制备方法
- 一种磺酸基修饰介孔材料负载杂多酸催化剂的制备方法及其在酯化反应中的应用
- (S)-1-(1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- (R)-1-(1-苯乙基)硫脲修饰的Cr-Anderson型杂多酸催化剂、制备方法及其应用
- 稀土改性杂多酸催化剂、其制备方法及生物柴油的制备方法