KNN膜形成用液体组合物及使用该液体组合物的KNN膜的形成方法与流程
knn膜形成用液体组合物及使用该液体组合物的knn膜的形成方法
技术领域
[0001]
本发明涉及一种不含铅、且能够形成致密的膜的knn膜形成用液体组合物及使用该液体组合物的knn膜的形成方法。在本说明书中,knn为铌酸钾钠((k,na)nbo3)的简称。
[0002]
本申请基于2018年6月22日在日本申请的专利申请2018-118737号,主张其优先权,并将其内容援用于此。
背景技术:
[0003]
作为搭载于致动器或超音波装置等称作mems(micro electro mechanical system:微机电系统)的装置的压电元件的压电体层,向来使用具有高压电特性的pzt(锆钛酸铅)。然而,在环保方面,要求开发压低铅的含量的压电材料。作为其中一种压电材料,已开发有由knn构成的压电材料。
[0004]
以往,由knn构成的压电材料的液体组合物包含:包含钾、钠及铌的金属络合物混合物、硅油与溶剂,相对于金属络合物混合物与溶剂的总量100容量份含有5容量份以下的硅油。该液体组合物,通过包含规定量的硅油,能够抑制形成压电陶瓷膜时的烧成工序中的热膨胀,来降低压电陶瓷膜的残留应力,而能够作为压电陶瓷膜形成用组合物使用(例如参考专利文献1)。
[0005]
以钾(k)、钠(na)、铌(nb)的各金属成为所期望的摩尔比的方式使这些金属络合物溶解、分散于溶剂,由此制备上述金属络合物混合物。而且,作为含k金属络合物,可举出例如2-乙基己酸钾、乙酸钾、乙酰丙酮酸钾、乙氧化钾等。作为含na金属络合物,可举出例如2-乙基己酸钠、乙酸钠、乙酰丙酮酸钠、乙氧化钠等。作为含nb金属络合物,可举出例如乙氧化铌、2-乙基己酸铌、五乙氧化铌等。并且,作为上述溶剂,可举出甲苯、二甲苯、辛烷、乙二醇、2-甲氧基乙醇、丁醇、乙醇、异丙醇、乙酸、水、等各种溶剂。
[0006]
专利文献1:日本专利公开2012-169467(摘要、第[0023]段、[0025]段)
[0007]
迄今为止,通过将这种液体组合物以化学溶液沉积(csd:chemical solution deposition)法涂布于基板的电极上并进行干燥、预烧后,进行烧成,由此形成knn膜。然而,使用专利文献1所示的选自甲苯、二甲苯、辛烷、乙二醇、2-甲氧基乙醇、丁醇、乙醇、异丙醇、乙酸及水的1种以上作为溶剂来形成knn膜时,存在不易获得致密的膜的问题。
技术实现要素:
[0008]
本发明目的在于提供一种不含铅、且能够形成致密的膜的knn膜形成用液体组合物及使用该液体组合物的knn膜的形成方法。
[0009]
本案发明人等发现,在csd法中,作为knn膜形成用液体组合物的溶剂,在液体组合物的金属化合物使用特定的羧酸盐,另外,对于溶剂也以特定的羧酸作为主溶剂时,在液体组合物的分解时放热反应峰会变大,而能够解决上述课题,由此完成本发明。
[0010]
本发明第1观点为knn膜形成用液体组合物,其包含:有机金属化合物与溶剂,所述
有机金属化合物包含有机钾化合物、有机钠化合物及有机铌化合物,其特征在于,所述有机钾化合物及所述有机钠化合物分别为由通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸的金属盐,所述有机铌化合物为烷氧化铌或由所述通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸的金属盐,所述溶剂中主溶剂为由所述通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸,且相对于所述液体组合物100质量%包含50质量%~90质量%。
[0011]
本发明第2观点为一种knn膜的形成方法,将基于第1观点的knn膜形成用液体组合物涂布于基板的电极上之后,以150℃以上且350℃以下的温度进行预烧而形成预烧膜,再将所述预烧膜以10℃/秒以上的速度升温并以600℃以上且800℃以下的温度进行烧成而形成结晶化的knn膜。
[0012]
本发明第1观点的knn膜形成用液体组合物中,通过在作为原料种类的金属化合物中使用特定的羧酸,而且对于溶剂也以该特定的羧酸作为主溶剂,由此在液体组合物的分解时放热反应峰会变大,使液体组合物一举被烧成,残留碳较少,而能够形成致密的knn膜。
[0013]
本发明第2观点的knn膜的形成方法中,涂布knn膜形成用液体组合物后,以规定的温度进行预烧而形成预烧膜,再将该预烧膜以10℃/秒以上的速度升温并以600℃以上且800℃以下的温度一体进行烧成。该方法中,对液体组合物的涂布膜以规定的温度进行预烧并以规定的升温速度进行烧成,由此液体组合物的分解时的放热反应峰会变大,使液体组合物一举被烧成,残留碳较少,而能够形成致密的knn膜。
附图说明
[0014]
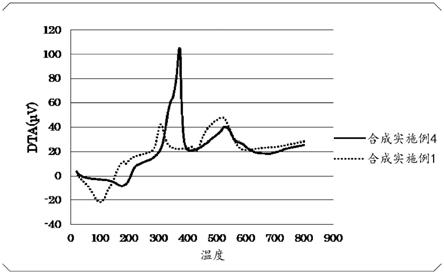
图1为表示将合成实施例4及合成比较例1的液体组合物在大气压下以10℃/秒从室温升温至800℃时的dta测量结果的图表。
具体实施方式
[0015]
对用于实施本发明的实施方式进行说明。
[0016]
[knn膜形成用液体组合物]
[0017]
本实施方式的knn膜形成用液体组合物包含有机金属化合物与溶剂,该有机金属化合物包含有机钾化合物、有机钠化合物及有机铌化合物。由该液体组合物所形成的knn膜是通过铌酸钾钠((k,na)nbo3)的钙钛矿型结构的有机金属化合物的复合氧化物所构成的。本实施方式的复合氧化物在a位包含钾(k)及钠(na),在b位包含铌(nb)。在该钙钛矿型的abo3型结构中,在a位,氧成12配位,且在b位,氧成六配位而形成八面体(octahedron),钾及钠位于该a位,铌则位于b位。
[0018]
本实施方式的有机金属化合物,即复合氧化物的金属摩尔比不特别限定,k:na:nb优选为x:1-x:y(其中,0.1≤x≤0.7、0.7≤y≤1.4)。另外,若y小于0.7,则nb源过少而有可能产生异相;若y超过1.4,则nb源过多而有可能产生异相。
[0019]
本实施方式的有机钾化合物及有机钠化合物分别为由通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸的金属盐。若n小于4,则膜不够致密;n超过8,则如后述使用羧酸作为主溶剂时,主溶剂会成为固体而不适合作为溶剂。n优选为6~8的范围。
[0020]
并且,本实施方式的有机铌化合物为烷氧化铌或由上述通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸的金属盐。
[0021]
由上述通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸,具体而言为以下表1所示的化合物。
[0022]
当有机钾化合物及有机钠化合物不是羧酸的金属盐,例如为烷氧化钾或烷氧化钠时,在由液体组合物形成膜的过程中会产生异相。但有机铌化合物不是羧酸的金属盐,而是烷氧化铌也无妨。
[0023]
烷氧化铌能够使用五乙氧基铌(别称乙氧基铌)等。
[0024]
[表1]
[0025][0026]
本实施方式的knn膜形成用液体组合物所含的溶剂中,主溶剂为由上述通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸。作为主溶剂的羧酸,相对于上述液体组合物100质量%含有50质量%~90质量%,优选含有70质量%~80质量%。主溶剂的羧酸若小于50质量%,则存在在液体组合物的制备中途产生沉淀的问题;若超过90质量%,则存在形成的膜过薄而导致生产性变差的问题。由上述通式所示的羧酸以外的溶剂可举出醇、乙酸、水等。
[0027]
在此,有机金属化合物的羧酸的金属盐若为例如2-乙基己酸钾、2-乙基己酸钠、2-乙基己酸铌等2-乙基己酸的金属盐,则从溶液中不会产生多种副产物观点优选主溶剂也为2-乙基己酸。即,构成金属盐的羧酸与主溶剂的羧酸优选是同一种。
[0028]
[knn膜形成用液体组合物的制备方法]
[0029]
将作为knn的金属源的上述有机钾化合物(k源)、有机钠化合物(na源)与有机铌化合物(nb源)溶解于包含上述主溶剂的溶剂、来制备本实施方式的knn膜形成用液体组合物。具体而言,首先向容器加入包含上述作为溶剂的羧酸的有机溶剂(包含能够以后述的减压蒸馏脱离的有机溶剂的安定剂等)与有机钠化合物,以130℃~170℃的油浴进行回流30分~60分钟而得到红褐色的悬浮液。对其添加有机钾化合物及有机铌化合物,以相同温度的油浴持续进行回流30分~60分钟而制备合成液。在此,以金属摩尔比(k:na:nb)成为x:1-x:y(其中,0.1≤x≤0.7、0.7≤y≤1.4)的方式分别秤量上述有机钾化合物(k源)、上述有机钠化合物(na源)及上述有机铌化合物(nb源)。
[0030]
接着进行减压蒸馏而从合成液中使溶剂脱离,从而去除有机溶剂及反应副产物。对所得溶液根据需要添加作为溶剂的羧酸、醇、水等,而将溶液稀释至钾、钠及铌的总量以金属氧化物换算为6质量%~20质量%。通过将所得稀释液以过滤器过滤而去除残留物,从而得到液体组合物。钾、钠及铌的总量以金属氧化物换算若小于6质量%,虽可获得良好的膜,但因膜厚过薄,直至获得期望的厚度为止生产性会变差。若超过20质量%,则在液体组合物中容易产生沉淀。
[0031]
[knn膜的形成方法]
[0032]
本实施方式的knn膜形成于基板上。该基板具有硅制或蓝宝石制的具有耐热性的基板本体。若为硅制基板本体,则在该基板本体上设置sio2膜,并在该sio2膜上设置由pt、tio
x
、ir、ru等具有导电性且不会与knn膜反应的材料所构成的下部电极。例如,能够将下部电极作成从基板本体侧依次具有tio
x
膜及pt膜的双层构造。上述tio
x
膜的具体例可举出tio2膜。而且,上述sio2膜是为了提升密接性而形成。pt膜例如通过溅镀法,朝(111)面取向而形成。
[0033]
在该下部电极的pt膜上通过csd法涂布上述液体组合物并进行预烧、烧成而形成knn膜。该液体组合物的涂布能够以通过旋转涂布、浸渍涂布、lsmcd(liquid source misted chemical deposition,液态源雾化化学沉积)法或静电喷涂法等,在进行预烧后以形成具有50nm以上且150nm以下的厚度的涂膜(凝胶膜)的方式来进行。预烧后的膜厚若小于50nm,则虽可获得良好的膜,但因膜厚过薄,存在直至获得期望的厚度为止生产性会变差的问题;若超过150nm,则在烧成后的knn膜中容易产生裂痕。
[0034]
涂布上述液体组合物后的预烧通过例如加热板或红外线急速加热炉(rta),在150℃以上且400℃以下,优选为200℃以上且350℃以下的温度进行。预烧温度若小于150℃,则存在不会形成凝胶状的问题。若超过400℃,则knn膜不易结晶化。并且,预烧之后预烧膜的厚度若小于50nm,则虽可获得良好的膜,但因膜厚过薄,存在直至获得期望的厚度为止生产性会变差的问题;若超过150nm,则在烧成后的knn膜中容易产生裂痕。
[0035]
制作预烧膜后,对此预烧膜进行烧成。该烧成通过在含氧(o2)环境中,将预烧膜通过rta(rapid thermal annealing,快速热退火),以10℃/秒以上的速度升温至600℃以上且800℃以下的温度,并保持0.5分钟以上5分钟以下的时间来进行。优选的升温速度为40
℃/秒以上且60℃/秒以下,优选的烧成温度为650℃以上且750℃以下。若升温速度小于10℃/秒、烧成温度小于600℃,则制作的knn膜的结晶化度不够充分,导致其密度降低。烧成温度若超过800℃,则基板等会发生损伤。
[0036]
实施例
[0037]
接着连同比较例一起来详细说明本发明的实施例。
[0038]
<合成实施例1>
[0039]
在烧瓶中以成为5:3:1:4的摩尔比的方式添加作为溶剂的α-甲基丁酸、作为添加剂的乙酸酐、作为有机钠化合物的α-甲基丁酸钠(na源)及作为安定剂的琥珀酸二甲酯而制备了悬浮液。接着将烧瓶内所得的悬浮液以150℃的油浴在150℃下进行回流30分钟。回流后,对经回流的液体添加作为有机钾化合物的α-甲基丁酸钾(k源)与作为有机铌化合物的五乙氧化铌(nb源),以150℃的油浴在150℃下进行回流30分钟而制备成合成液。在此,上述α-甲基丁酸钾(k源)、上述α-甲基丁酸钠(na源)及上述五乙氧化铌(nb源)以金属摩尔比(k:na:nb)成50:50:100的方式分别秤量。回流后,对经回流的液体添加水与乙醇,再次以150℃的油浴在150℃下进行回流30分钟。回流后,用真空发生器进行减压蒸馏至0.015mpa。由此去除未反应产物,而得到作为knn前驱物的液体组合物。对液体组合物添加作为溶剂的α-甲基丁酸而将液体组合物稀释成金属氧化物(钾、钠及铌)的浓度在该液体组合物中占10质量%。相对于经稀释的液体组合物100质量%,作为主溶剂的α-甲基丁酸的含有比例为70质量%。将稀释液用过滤器过滤而去除残留物。
[0040]
将制备合成实施例1的液体组合物的有机金属化合物(有机钾化合物、有机钠化合物及有机铌化合物)的种类及液体组合物中的金属摩尔比(k:na:nb),以及主溶剂的种类、主溶剂为羧酸的情况下上述通式c
n
h
2n+1
cooh中的n的数目及上述主溶剂相对于液体组合物100质量%的含量(质量%)分别示于表2。
[0041]
[表2]
[0042][0043]
<合成实施例2~8及合成比较例1~5>
[0044]
使用表2所示的作为有机金属化合物的有机钾化合物、有机钠化合物及有机铌化合物,以成为表2所示金属摩尔比的方式秤量这些有机金属化合物,并如表2所示变更主溶剂的含量。除此之外以与合成实施例1同样的方式,分别制备成合成实施例2~8及合成比较例1~5的液体组合物。合成比较例4中,使用烷氧化钾作为有机钾化合物。并且,合成比较例5中,使用烷氧化钠作为有机钠化合物。
[0045]
<实施例1>
[0046]
接着,在用于评价的基板上,使用合成实施例1中所得的液体组合物形成knn膜。基板使用了4英寸si基板。si基板通过热氧化形成了500nm的氧化膜。通过溅镀法在氧化膜上堆积20nm的ti,并在其上通过溅镀法形成了厚度100nm的(111)取向的pt下部电极。在所得基板上滴下0.5ml的合成实施例1中所得的液体组合物,以5000rpm进行旋转涂布15秒,并将经旋转涂布的液体进行了干燥。进而,以300℃的加热板进行了预烧5分钟。之后,用rta以700℃、氧气气氛、升温速度50℃/秒、保持时间1分钟进行了烧成。由此在下部电极上形成了实施例1的knn膜。
[0047]
将获得实施例1中使用的液体组合物的合成实施例的种类(合成实施例1)、预烧温度、升温速度、烧成温度及保持时间分别示于表3。
[0048]
[表3]
[0049][0050]
<实施例2~12及比较例1~9>
[0051]
使用表3所示的合成实施例2~9及合成比较例1~5、合成实施例7中所得的液体组合物,以与实施例1同样方式,分别将液体组合物进行旋转涂布并加以干燥后,如表3所示般变更了预烧温度、升温速度、烧成温度及保持时间。除此之外,以与实施例1同样的方式在下部电极上形成了实施例2~13及比较例1~9的knn膜。另外,比较例2中,由于在液体中产生沉淀,且比较例3中,由于液体组合物无法均匀涂布,因此分别无法形成膜。
[0052]
<比较试验1>
[0053]
在氧化铝秤盘中分别采取20mg合成实施例4与合成比较例1中所合成的液体组合物,分别使用热重量差示热分析装置(tg-dta)(mac science公司制,tg-dta),将这些液体在大气压下、以10℃/秒的速度从室温升温至800℃,进行了差示热分析。将这些测量结果示于图1。
[0054]
<评价结果1>
[0055]
由图1明显可知,与合成比较例1的液体组合物相比,合成实施例4的液体组合物在380℃附近急剧地发生因分解与氧化所引起的放热反应。并且,除合成实施例4中使用的羧酸以外,使用通式c
n
h
2n+1
cooh(其中,4≤n≤8)所表示的羧酸时,与合成实施例4同样地发生了急剧的放热反应。
[0056]
<比较试验2>
[0057]
考察了实施例1~13及比较例1、比较例4~9中所得的20种knn膜的致密性。该knn膜的致密性通过测量knn膜的密度来考察。具体而言,以sem观察18种knn膜的剖面,并对其剖面影像进行影像解析而算出膜的面积及膜中的空隙部分的面积,再通过进行[(膜的面积-空隙部分的面积)/膜的面积]
×
100的计算而算出了膜密度(%)。当膜密度超过98%时判定为“优良”;处于90%~97%的范围时判定为“良好”;小于90%时则判定为“不良”。
[0058]
<评价结果2>
[0059]
由表3明显可知,比较例1中,knn膜的形成条件处于以150℃以上且350℃以下的温度进行预烧而形成预烧膜,再将所述预烧膜以10℃/秒以上的速度升温并以600℃以上且800℃以下的温度进行烧成的条件范围内,但未使用通式c
n
h
2n+1
cooh(其中,4≤n≤8)所示的羧酸的金属盐,且未使用通式c
n
h
2n+1
cooh(其中,4≤n≤8)所示的羧酸的主溶剂。比较例4~5中,knn膜的形成条件处于以150℃以上且350℃以下的温度进行预烧而形成预烧膜,再将所述预烧膜以10℃/秒以上的速度升温并以600℃以上且800℃以下的温度进行烧成的条件范围内,但有机钾化合物与有机钠化合物中的任一个均未使用通式c
n
h
2n+1
cooh(其中,4≤n≤8)所示的羧酸的金属盐。因此,认为在比较例1、比较例4~5中,在升温中未发生急剧的放热反应,残留碳较多而未形成致密的膜。
[0060]
比较例6~7中,使用与实施例7及10~13同样的由合成实施例7所得的液体组合物来进行了试验,但knn膜的形成条件处于以150℃以上且350℃以下的温度进行预烧而形成预烧膜,再将所述预烧膜以10℃/秒以上的速度升温并以600℃以上且800℃以下的温度进行烧成的条件范围以外。其结果,无法获得致密的knn膜。
[0061]
另一方面,实施例1~13中,作为主溶剂使用通式c
n
h
2n+1
cooh(其中,4≤n≤8)所示的羧酸,且以150℃以上且350℃以下的温度进行预烧而形成预烧膜,再将所述预烧膜以10℃/秒以上的速度升温并以600℃以上且800℃以下的温度进行烧成。因此,在升温中发生了急剧的放热反应,由此获得了致密的膜。认为这是因为,由于在升温中发生急剧的放热反应,使残留碳减少,而使膜变得更为致密。
[0062]
产业上的可利用性
[0063]
由本发明的knn膜形成用液体组合物及knn膜的制造方法所得的knn膜能够使用于致动器、超音波装置、振动发电元件、热释电传感器、喷墨头、自动对焦等mems应用的压电体膜。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1