一种治疗痛风性关节炎的片剂的制备方法及检测方法与流程
本发明涉及药物领域,特别涉及一种治疗痛风性关节炎的片剂的制备方法及检测方法。
背景技术:
痛风性关节炎是由于嘌呤代谢紊乱,尿酸产生过多和(或)尿酸排泄减少,血尿酸水平持续增高导致尿酸盐结晶沉积于关节及软组织所致的一组代谢性疾病。其主要临床表现为反复发作的急性痛风性关节炎、慢性痛风性关节炎、痛风石、痛风性肾病,严重者出现关节畸形及功能障碍。
临床对痛风性关节炎的认识:阳气者,精则养神,柔则养筋。阳气是机体功能的基础。损之,耗之,则气虚阳弱,导致脏腑功能低下,引发诸病。损脾阳虚,则脾主运化失司,水湿停聚,或招致外湿,而致醸痰、淤浊、成瘀、化毒;损肾阳致虚,则开阖失司,水道不利,所以,痛风的发病首先是脏腑之气不足,功能失调,导致痰湿浊瘀蕴毒于血,机体泻毒不力,日久留滞脏腑组织关节之隙,或发关节肿痛,或发脏腑绞痛,甚或脏腑衰竭。
但是目前的药物缺乏有效的鉴定方法,因此,提供一种专属性强、检测准确的痛风药物的鉴定方法具有重要的现实意义。
技术实现要素:
有鉴于此,本发明提供一种治疗痛风性关节炎的片剂的制备方法及检测方法。该制备方法以药效筛选工艺为基础,使用常规提取设备,更好的保留复方中有效成分或药效成分,工艺中没有用到有毒的有机溶剂,无污染,有利于保护环境,适于工业化生产。该检测方法中通过对样品处理方法的筛选,展开剂体系的选择,使得鉴别专属性好,而且方法经济适用、结果快速,解决了之前桂枝和五加皮薄层鉴别缺乏专属性的缺陷。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种治疗痛风性关节炎的片剂的制备方法,包括如下步骤:
步骤1:水提挥发油:取桂枝250质量份、海风藤500质量份、野菊花500质量份,粉碎,加6~12倍量水提取1.5~6h,收集挥发油;
步骤2:挥发油包合:取β-环糊精2.30~3.10质量份在20℃~35℃下研磨包合15~30min,过滤,干燥,得挥发油包合物;
步骤3:水提液浓缩与醇沉:收集步骤1提取挥发油后的水提液滤过,减压浓缩至相对密度1.10~1.15(60℃)的稠膏,放置至20~30℃,与95%乙醇混合使含醇量达55~85%,搅拌8~30min,20~30℃静置8~24h,取上清液,滤过,收集醇沉上清液;
步骤4:醇提:取黄芪750质量份、五加皮500质量份、浙贝母250质量份、刘寄奴500质量份、秦皮500质量份,加60~90%乙醇回流提取两次,第一次加6~12倍量提取1.5~2.5h,第二次加6~12倍量提取1~2h,滤过,合并滤液,得乙醇提取液;
步骤5:回收乙醇与浓缩:将步骤3制得的醇沉上清液、步骤4制得的乙醇提取液合并,减压回收乙醇,浓缩至相对密度1.20~1.25(60℃)的稠膏;
步骤6:干燥、粉碎:取步骤5制得的浸膏,干燥,粉碎,过80~100目筛,得提取干浸膏粉;
步骤7:制粒与干燥:取步骤6制得的所述提取干浸膏粉加糊精90~120质量份,混匀,以85~95%乙醇为粘合剂,湿法制粒,干燥,整粒,制得颗粒;
步骤8:压片:取步骤7制得的所述颗粒,与5.30~6.00质量份的硬脂酸镁、步骤2制得的挥发油包合物混合得混合物料,压片;
步骤9:薄膜包衣:取包薄膜衣粉16~20质量份加65~85%乙醇配置成混悬溶液,薄膜包衣。
在本发明的一些具体实施方案中,所述制备方法步骤3中所述减压浓缩的压强为-0.06mpa~0.08mpa。
在本发明的一些具体实施方案中,所述制备方法步骤5中所述浓缩的压强为-0.06mpa~-0.08mpa。
在本发明的一些具体实施方案中,所述制备方法步骤6中所述干燥的压强为-0.08~0.09mpa,所述干的燥温度为60~80℃。
在本发明的一些具体实施方案中,所述制备方法步骤8中所述混合的时间为15~30min;所述混合物料的水份<5.0%;压片控制硬度>6kg。
本发明还提供了所述制备方法制得的片剂。
本发明还提供了所述制备方法制得的片剂的检测方法,包括如下步骤:
步骤1:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研磨,与10~20倍体积量甲醇混合,超声处理25~40min,滤过,滤液蒸干,残渣加水5~10倍体积量至溶解,用水饱和的正丁醇萃取2~4次,每次10~20倍体积量,合并正丁醇萃取液,用1%氢氧化钠溶液洗涤2~4次,每次2~8倍体积量,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1~3ml使溶解,作为供试品溶液;
按供试品溶液的制备方法,制成黄芪阴性供试品溶液;
取黄芪对照药材1~3g,按供试品溶液的制备方法制成对照药材溶液;
取黄芪甲苷对照品,加甲醇制成每1ml含0.8~1.5mg的黄芪甲苷对照品溶液;
按照薄层色谱法,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,即为合格;
步骤2:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研磨,加2~8倍体积量甲醇,加热回流8~15min,放冷,滤过,滤液作为供试品溶液;
按供试品溶液的制备方法,制成秦皮阴性供试品溶液;
取秦皮对照药材1~3g,按供试品溶液的制备方法制成秦皮对照药材溶液;
取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含1.5~2.5mg的混合溶液,作为对照品溶液;
按照薄层色谱法试验,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,即为合格;
步骤3:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研细,加浓氨试液3~6ml与所述片剂10~20倍体积量二氯甲烷,加热回流0.5~1.5h,放冷,滤过,滤液用0.1~0.2mol/l的盐酸溶液振摇提取1~3次,每次2~8倍体积量,合并提取液,用浓氨调节ph到9~11用二氯甲烷振摇提取1~3次,每次2~8倍体积量,合并二氯甲烷液,蒸干,加无水乙醇1ml溶解,作为供试品溶液;
按供试品溶液的制备方法,制成浙贝母阴性供试品溶液;
取浙贝母对照药材3~6g,按供试品溶液的制备方法制成浙贝母对照药材溶液;
另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含1.5~2.5mg的混合溶液,作为对照品溶液;
按照薄层色谱法试验,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点,即为合格;
步骤4:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研细,加二氯甲烷10~20倍体积量,超声处理25~40min,滤过,滤液蒸干,残渣加二氯甲烷1~3ml使溶解,作为供试品溶液;
按供试品溶液的制备方法,制成五加皮阴性供试品溶液;
取五加皮对照药材0.5~2g,按供试品溶液的制备方法制成五加皮对照药材溶液;
按照薄层色谱法试验,供试品、对照药材在薄层色谱图相同位置上,显相同颜色的斑点,即为合格;
步骤5:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研细,加二氯甲烷8~15倍体积量,超声25~40min,滤过,加入等体积1%naoh溶液萃取1~4次,合并萃取液,加盐酸酸化至ph值为2~4,再用等体积二氯甲烷萃取1~4次,合并萃取液,挥去二氯甲烷,残渣加甲醇0.5ml使溶解,作为供试品溶液;
按供试品溶液的制备方法,制成桂枝阴性供试品溶液;
取桂枝对照药材1~3g,按供试品溶液的制备方法制成桂枝对照药材溶液;
取肉桂酸对照品,加乙醇制成每1ml含0.8~1.6mg的溶液,作为对照品溶液;
按照薄层色谱法试验,供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点,即为合格。
本发明还提供了所述制备方法制得的片剂的检测方法,包括如下步骤:
步骤1:取如权利要求1至5任一项所述制备方法制得的片剂(0.65g/片),除去包衣,研磨,与40~60ml甲醇混合,超声处理25~40min,滤过,滤液蒸干,残渣加水20~40ml至溶解,用水饱和的正丁醇萃取2~4次,每次40~60ml,合并正丁醇萃取液,用1%氢氧化钠溶液洗涤2~4次,每次15~30ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1~3ml使溶解,作为供试品溶液;
按供试品溶液的制备方法,制成黄芪阴性供试品溶液;
取黄芪对照药材1~3g,按供试品溶液的制备方法制成对照药材溶液;
取黄芪甲苷对照品,加甲醇制成每1ml含0.8~1.5mg的黄芪甲苷对照品溶液;
按照薄层色谱法(2010年版药典附录ⅵb),供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,即为合格;
步骤2:取如权利要求1至5任一项所述制备方法制得的片剂(0.65g/片),除去包衣,研磨,加甲醇15~30ml,加热回流8~15min,放冷,滤过,滤液作为供试品溶液;
按供试品溶液的制备方法,制成秦皮阴性供试品溶液;
取秦皮对照药材1~3g,按供试品溶液的制备方法制成秦皮对照药材溶液;
取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含1.5~2.5mg的混合溶液,作为对照品溶液;
按照薄层色谱法(2010年版药典附录ⅵb)试验,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,即为合格;
步骤3:取如权利要求1至5任一项所述制备方法制得的片剂(0.65g/片),除去包衣,研细,加浓氨试液3~6ml与二氯甲烷45~60ml,加热回流0.5~1.5h,放冷,滤过,滤液用0.1~0.2mol/l的盐酸溶液振摇提取1~3次,每次10~30ml,合并提取液,用浓氨调节ph到9~11用二氯甲烷振摇提取1~3次,每次10~30ml,合并二氯甲烷液,蒸干,加无水乙醇1ml溶解,作为供试品溶液;
按供试品溶液的制备方法,制成浙贝母阴性供试品溶液;
取浙贝母对照药材3~6g,按供试品溶液的制备方法制成浙贝母对照药材溶液;
另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含1.5~2.5mg的混合溶液,作为对照品溶液;
按照薄层色谱法(2010年版药典附录ⅵb)试验,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点,即为合格;
步骤4:取如权利要求1至5任一项所述制备方法制得的片剂(0.65g/片),除去包衣,研细,加二氯甲烷55~70ml,超声处理25~40min,滤过,滤液蒸干,残渣加二氯甲烷1~3ml使溶解,作为供试品溶液;
按供试品溶液的制备方法,制成五加皮阴性供试品溶液;
取五加皮对照药材0.5~2g,按供试品溶液的制备方法制成五加皮对照药材溶液;
按照薄层色谱法(2015年版《中国药典》附录ⅵb)试验,供试品、对照药材在薄层色谱图相同位置上,显相同颜色的斑点,即为合格;
步骤5:取如权利要求1至5任一项所述制备方法制得的片剂(0.65g/片),除去包衣,研细,加二氯甲烷35~50ml,超声25~40min,滤过,加入等体积1%naoh溶液萃取1~4次,合并萃取液,加盐酸酸化至ph值为2~4,再用等体积二氯甲烷萃取1~4次,合并萃取液,挥去二氯甲烷,残渣加甲醇0.5ml使溶解,作为供试品溶液;
按供试品溶液的制备方法,制成桂枝阴性供试品溶液;
取桂枝对照药材1~3g,按供试品溶液的制备方法制成桂枝对照药材溶液;
取肉桂酸对照品,加乙醇制成每1ml含0.8~1.6mg的溶液,作为对照品溶液;
按照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点,即为合格;
步骤6:含量测定,每片含黄芪以黄芪甲苷(c41h68o14)计,不少于0.16mg。
在本发明的一些具体实施方案中,所述检测方法步骤1中的薄层色谱法具体为:分别吸取供试品溶液、阴性供试品溶液、对照药材溶液和黄芪甲苷对照品溶液4种溶液各4~10μl,点于同一硅胶g薄层板上,以氯仿:甲醇:水(13~10:7~4:2~0.4)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,分别在日光下检视;
步骤2中所述薄层色谱法具体为:吸取供试品溶液、阴性供试品溶液秦皮对照药材溶液和对照品溶液4种溶液各1~6μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-甲酸(6~4:1~05:0.5~0.1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视;
步骤3中所述薄层色谱法具体为:吸取供试品溶液、阴性供试品溶液浙贝母对照药材溶液和对照品溶液4种溶液各3~8μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-甲醇-浓氨试液(17~13:2~0.8:1~0.4)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液;
步骤4中所述薄层色谱法具体为:吸取供试品溶液、阴性供试品溶液和五加皮对照药材溶液3种溶液各1~5μl,分别点于同一硅胶g薄层板上,以石油醚(60~90℃)-丙酮-异丙醇-甲酸(12~6:2~1:0.5~0.2:0.1~0.04)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,在日光下检视;
步骤5中所述薄层色谱法具体为:吸取供试品溶液、阴性供试品溶液、桂枝对照药材溶液和对照品溶液4种溶液各5~10μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(5~3:5~3:0.1~0.02)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。
在本发明的一些具体实施方案中,所述检测方法步骤6所述含量测定具体按照以下步骤a~步骤c进行:
a、色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;流动相:乙腈-水(25~35:75~65)为流动相;流速:0.8~1.2ml·min-1;柱温:30℃;气体流速:1.5~2.7l/min;漂移管温度:50~105℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
b、溶液制备:
供试品溶液的制备:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研磨,加10~20倍体积量甲醇,超声提取20~40min,滤过,回收溶剂并浓缩至干,残渣加水2~4倍体积量,微热使溶解,用水饱和的正丁醇振摇提取2~4次,每次10~20倍体积量,合并正丁醇提取液,用氨试液充分洗涤1~3次,每次10~20倍体积量,弃去氨液,正丁醇液蒸干,残渣加水8~15ml使溶解,放冷,通过d101型大孔吸附树脂柱,以水10~15倍体积量洗脱,弃去水液,再用30%~40%乙醇5~12倍体积量洗脱,弃去洗脱液,继用70%~85%乙醇20~30倍体积量洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至5ml或10ml量瓶中,加甲醇至刻度,摇匀;
黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品,加甲醇分别制成每1ml含0.25~0.30mg、每1ml含0.50~0.60mg的对照品溶液;
c、测定法分别精密吸取对照品溶液与供试品溶液各5~10μl,注入液相色谱仪,测定。
在本发明的一些具体实施方案中,所述检测方法步骤6所述含量测定具体按照以下步骤a~步骤c进行:
a、色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;流动相:乙腈-水(25~35:75~65)为流动相;流速:0.8~1.2ml·min-1;柱温:30℃;气体流速:1.5~2.7l/min;漂移管温度:50~105℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
b、溶液制备:
供试品溶液的制备:取如权利要求1至5任一项所述制备方法制得的片剂,除去包衣,研磨,称取3.5~4.5g,精密称定,加甲醇35~45ml,超声提取20~40min,滤过,回收溶剂并浓缩至干,残渣加水8~12ml,微热使溶解,用水饱和的正丁醇振摇提取2~4次,每次35~45ml,合并正丁醇提取液,用氨试液充分洗涤1~3次,每次35~45ml,弃去氨液,正丁醇液蒸干,残渣加水8~15ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水40~50ml洗脱,弃去水液,再用30%~40%乙醇20~40ml洗脱,弃去洗脱液,继用70%~85%乙醇70~90ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至5ml或10ml量瓶中,加甲醇至刻度,摇匀;
黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品,加甲醇分别制成每1ml含0.25~0.30mg、每1ml含0.50~0.60mg的对照品溶液;
c、测定法分别精密吸取对照品溶液与供试品溶液各5~10μl,注入液相色谱仪,测定。
本发明中所述“倍体积量”是相对于本发明提供的治疗痛风性关节炎的片剂。即表示以g/ml计,所述片剂与添加的溶剂的质量体积比。
本发明公开了一种芪桂痛风片的制备方法和质量控制方法,所述制剂主要由下述原料制备而得:黄芪,五加皮,刘寄奴,浙贝母,秦皮,野菊花,海风藤,桂枝;其制备方法为:取野菊花、海风藤、桂枝药材,水蒸气蒸馏提取挥发油,水提液滤过,滤液减压浓缩至稠膏,加入适量乙醇,静置过夜,滤过,得醇沉上清液,备用;挥发油用β-环糊精包合,过滤,干燥,得挥发油包合物;黄芪、五加皮、浙贝母、刘寄奴、秦皮药材加乙醇回流提取,滤过,得乙醇提取液,备用;将醇沉上清液、乙醇提取液合并,浓缩至稠膏,干燥,粉碎加入糊精制粒,再加挥发油包合物和硬脂酸镁混匀,制成片剂;本发明提供的质量控制方法包括黄芪、浙贝母、秦皮、桂枝和五加皮的薄层鉴别,黄芪甲苷的高效液相色谱含量测定及其限度。
本发明提供的质量检测方法,是通过具有创造性试验筛选后得到,检测方法中通过对样品处理方法的筛选,展开剂体系的选择,使得鉴别专属性好,而且方法经济适用、结果快速,解决了之前桂枝和五加皮薄层鉴别缺乏专属性的缺陷。含量测定方法中通过对供试品处理方法的优选,增加了黄芪甲苷的含量测定,使得含量测定方法可以很有效的对产品进行质量控制,能满足实际生产的要求。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
图1示实施例制备的芪桂痛风片中黄芪药材的薄层鉴别的tlc图;其中,泳道1、黄芪阴性制剂;泳道2、制剂20150101;3、泳道黄芪甲苷对照品;泳道4、制剂20150102;泳道5、黄芪对照药材;泳道6、制剂20150201;
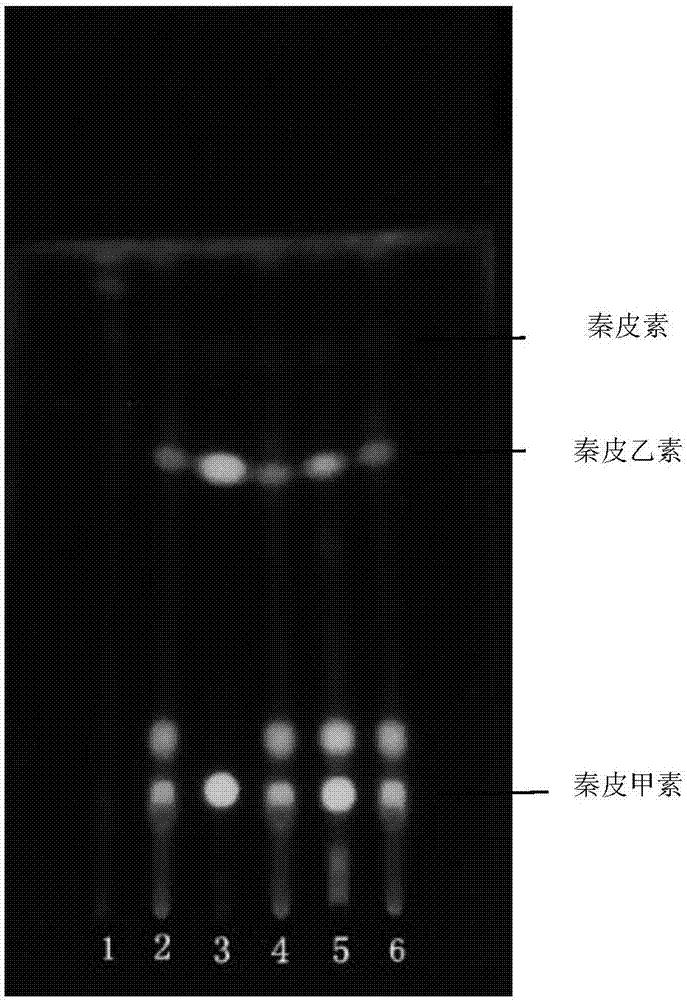
图2示实施例制备的芪桂痛风片中秦皮药材的薄层鉴别的tlc图;其中,泳道1、秦皮阴性制剂;泳道2、制剂20150101;泳道3、秦皮素、秦皮甲素、秦皮乙素混合对照品;泳道4、制剂20150102;泳道5、秦皮对照药材;泳道6、制剂20150201;
图3示实施例制备的芪桂痛风片中药材浙贝母薄层鉴别的tlc图;其中,泳道1、浙贝母阴性制剂;泳道2、制剂20150101;泳道3、贝母素甲、贝母素乙混合对照品;泳道4、制剂20150102;泳道5、浙贝母对照药材;6、制剂20150201;
图4示实施例制备的芪桂痛风片中五加皮药材的薄层鉴别的tlc图;其中,泳道1、五加皮阴性制剂;泳道2、制剂20150101;泳道3、五加皮对照药材;泳道4、制剂20150102;泳道5、五加皮对照药材;泳道6、制剂20150201;
图5示实施例制备的芪桂痛风片中桂枝药材的薄层鉴别的tlc图;其中,泳道1、桂枝阴性制剂;泳道2、制剂20150101;泳道3、肉桂酸对照品;泳道4、制剂20150102;泳道5、桂枝对照药材;泳道6、制剂20150201;
图6示芪桂痛风片中黄芪甲苷的线性关系图。
具体实施方式
本发明公开了一种治疗痛风性关节炎的片剂的制备方法及检测方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
本发明提供了一种芪桂痛风片的制备方法。
本发明提供了一种芪桂痛风片的质量控制方法。
本方面提供的一种芪桂痛风片的制备方法,其特征在于,由黄芪、桂枝、秦皮、五加皮、野菊花、海风藤、刘寄奴、浙贝母、β-环糊精、糊精、硬脂酸镁、薄膜包衣粉,制成片剂。
所述芪桂痛风片采用如下制备方法获得:
(1)水提挥发油:取桂枝250g、海风藤500g、野菊花500g,粉碎成最粗粉,加6~12倍量水提取1.5~6小时,收集挥发油;
(2)挥发油包合:用β-环糊精2.30~3.10g在20℃~35℃下研磨包合15~30min,过滤,干燥,得挥发油包合物;
(3)水提液浓缩与醇沉:提取挥发油后的水提液滤过,减压浓缩至相对密度1.10~1.15(60℃)的稠膏,放置至室温,边加入乙醇边搅拌使含醇量达55~85%,继续搅拌8~30min,室温(20~30℃)静置8~24h,取上清液,滤过,即得醇沉上清液,备用;
(4)醇提:黄芪750g、五加皮500g、浙贝母250g、刘寄奴500g、秦皮500g,加60~90%乙醇回流提取两次,第一次加6~12倍量提取1.5~2.5小时,第二次加6~12倍量提取1~2小时,滤过,合并滤液,得乙醇提取液,备用;
(5)回收乙醇与浓缩:将醇沉上清液、乙醇提取液合并,减压回收乙醇,浓缩至相对密度1.20~1.25(60℃)的稠膏,备用;
(6)干燥、粉碎:取上述浸膏,置真空干燥器内干燥,粉碎,过80~120目筛,得提取干浸膏粉,置不锈钢桶内,备用;
(7)制粒与干燥:取上述干浸膏粉加糊精90~120g,混匀,以85~95%乙醇为粘合剂,湿法制粒,干燥,整粒,备用;
(8)压片:取上述干燥好的颗粒,再加硬脂酸镁5.30~6.00g、挥发油包合物,混匀,制成本品1000片;
(9)薄膜包衣:取包薄膜衣粉16~20g加65~85%乙醇配置成混悬溶液,进行薄膜包衣,即得。
在本发明的一些具体实施方案中,所述的制备方法,其中步骤(3)中滤液减压浓缩的压强是-0.06mpa~0.08mpa。
在本发明的一些具体实施方案中,所述的制备方法,其中步骤(5)中药液减压浓缩的压强是-0.06mpa~-0.08mpa。
在本发明的一些具体实施方案中,所述的制备方法,其中步骤(6)中置真空干燥器内干燥粉碎的压强为-0.08~0.09mpa,干燥温度为60~80℃。
在本发明的一些具体实施方案中,所述的制备方法,其中步骤(6)中的混匀是在混合机中混合15~30分钟,测定混合物料的水份小于3.0~5.0%,压片控制硬度>6~8kg。
另一方面,本发明还提供了一种制备所述芪桂痛风片的检测方法,其中:
(1)将按原料比例制备好的芪桂痛风片,取5~10片,除去包衣,研细,加甲醇40~60ml,超声处理25~40min,滤过,滤液蒸干,残渣加水20~40ml使溶解,用水饱和的正丁醇萃取2~4次,每次40~60ml,合并正丁醇液,用1%氢氧化钠溶液洗涤2~4次,每次15~30ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1~3ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺黄芪阴性对照品溶液;取黄芪对照药材1~3g,按供试品溶液的制备方法制成对照药材溶液;取黄芪甲苷对照品,加甲醇制成每1ml含0.8~1.5mg的黄芪甲苷对照品溶液;照薄层色谱法(2010年版药典附录ⅵb),分别吸取上述4种溶液各4~10μl,点于同一硅胶g薄层板上,以氯仿:甲醇:水(13~10:7~4:2~0.4)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,分别在日光下检视;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(2)将按原料比例制备好的芪桂痛风片,取4~10片,除去包衣,研细,加甲醇15~30ml,加热回流8~15min,放冷,滤过,滤液作为供试品溶液;按原料比例的制备方法,制成缺秦皮阴性对照品溶液;取秦皮对照药材1~3g,按供试品溶液的制备方法制成秦皮对照药材溶液;取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含1.5~2.5mg的混合溶液,作为对照品溶液;照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各1~6μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-甲酸(6~4:1~05:0.5~0.1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(3)将按原料比例制备好的芪桂痛风片,取8~15片,除去包衣,研细,加浓氨试液3~6ml与二氯甲烷45~60ml,加热回流0.5~1.5h,放冷,滤过,滤液用0.1~0.2mol/l的盐酸溶液振摇提取1~3次,每次10~30ml,合并提取液,用浓氨调节ph到9~11用二氯甲烷振摇提取1~3次,每次10~30ml,合并二氯甲烷液,蒸干,加无水乙醇1ml溶解,作为供试品溶液;按原料比例的制备方法,制成缺浙贝母阴性对照品溶液;取浙贝母对照药材3~6g,按供试品溶液的制备方法制成浙贝母对照药材溶液;另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含1.5~2.5mg的混合溶液,作为对照品溶液;照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各3~8μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-甲醇-浓氨试液(17~13:2~0.8:1~0.4)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点;
(4)将按原料比例制备好的芪桂痛风片,取5~10片,除去包衣,研细,加二氯甲烷55~70ml,超声处理25~40min,滤过,滤液蒸干,残渣加二氯甲烷1~3ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺五加皮阴性对照品溶液;取五加皮对照药材0.5~2g,按供试品溶液的制备方法制成五加皮对照药材溶液;照薄层色谱法(2015年版《中国药典》附录ⅵb)试验,吸取上述两种溶液各1~5μl,分别点于同一硅胶g薄层板上,以石油醚(60~90℃)-丙酮-异丙醇-甲酸(12~6:2~1:0.5~0.2:0.1~0.04)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,在日光下检视;供试品、对照药材在薄层色谱图相同位置上,显相同颜色的斑点;
(5)将按原料比例制备好的芪桂痛风片,取5~10片,除去包衣,研细,加二氯甲烷35~50ml,超声25~40min,滤过,加入等体积1%naoh溶液萃取1~4次,合并萃取液,加盐酸酸化至ph值为2~4,再用等体积二氯甲烷萃取1~4次,合并萃取液,挥去二氯甲烷,残渣加甲醇0.5ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺桂枝阴性对照品溶液;取桂枝对照药材1~3g,按供试品溶液的制备方法制成桂枝对照药材溶液;取肉桂酸对照品,加乙醇制成每1ml含0.8~1.6mg的溶液,作为对照品溶液;照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各5~10μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(5~3:5~3:0.1~0.02)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点;
(6)含量测定:具体按照以下步骤a~步骤d进行:
a、色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;流动相:乙腈-水(30~35:70~65)为流动相;流速:0.8~1.2ml·min-1;柱温:30℃;气体流速:1.5~2.7l/min;漂移管温度:50~105℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
b、溶液制备:供试品溶液的制备:取重量差异项下的本品8~15片,除去包衣,研细,称取3.5~4.5g,精密称定,置索氏提取器中,加甲醇35~45ml,冷浸过夜,再加甲醇适量,加热回流3~5小时,提取液回收溶剂并浓缩至干,残渣加水8~12ml,微热使溶解,用水饱和的正丁醇振摇提取2~4次,每次35~45ml,合并正丁醇提取液,用氨试液充分洗涤1~3次,每次35~45ml,弃去氨液,正丁醇液蒸干,残渣加水4~6ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水40~50ml洗脱,弃去水液,再用30%~40%乙醇30ml洗脱,弃去洗脱液,继用70%乙醇70~90ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至5ml或10ml量瓶中,加甲醇至刻度,摇匀,即得。
黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品适量,加甲醇分别制成每1ml含0.25~0.30mg、每1ml含0.50~0.60mg的对照品溶液,即得。
专属性试验:按照步骤a所述的色谱条件,分别精密吸取供试品溶液、黄芪阴性对照溶液、对照品溶液各10μl,注入高效液相色谱仪,测定,结果表明黄芪阴性制剂无干扰,专属性好。
c、测定法分别精密吸取对照品溶液与供试品溶液各5~10μl,注入液相色谱仪,测定,即得。
优选的,每片含黄芪以黄芪甲苷(c41h68o14)计,不少于0.16mg。
全方黄芪补气,有益气行血之妙,为君;五加皮补肝肾、强筋骨、祛风湿,桂枝温阳气、通经脉、利关节。两药一助黄芪补虚扶阳,一祛风寒湿邪以通络止痛,共为臣药;三药均有消除水饮之功,合用又能利水以消痰。佐以浙贝母消痰散结、刘寄奴活血化瘀,通经止痛,海风藤、秦皮祛风湿、通经络止痛。共凑活血、化痰、祛风湿、通经络止痛之效。野菊花性微寒,善解毒,既增秦皮、浙贝母清热解毒之用;又制诸药温燥之性,为佐使。全方益气助阳、祛湿、化痰、活血化瘀、通络止痛,温补而不燥烈,祛邪而不伤正,故有扶正祛邪,通络止痛之效。
本发明制得的片剂熔扶正祛邪、清热祛湿、化痰散结、活血逐瘀、通络止痛之功效于一炉,并寓有扶正固本、标本兼治原则,组方遣药合理,临证疗效显著,是治疗脏腑气弱、痰瘀阻滞、经络不通所致的慢性痛风性关节炎(症见关节肿痛、屈伸不利等)之良剂。
本发明提供的质量检测方法,是通过具有创造性试验筛选后得到,鉴别方法中通过对样品处理方法的筛选,展开剂体系的选择,使得鉴别专属性好,而且方法经济适用、结果快速,解决了之前桂枝和五加皮薄层鉴别缺乏专属性的缺陷。含量测定方法中通过对供试品处理方法的优选,增加了黄芪甲苷的含量测定,使得含量测定方法可以很有效的对产品进行质量控,能满足实际生产的要求。
本发明提供的治疗痛风性关节炎的片剂的制备方法及检测方法中所用原料及试剂均可由市场购得。
下面结合实施例,进一步阐述本发明:
实施例1挥发油提取工艺的考察
本项目采用正交设计对工艺条件进行了优化,分别考察药材粒度、加水量、煎煮时间三因素,用正交表l9(33)进行试验,方案见表1,实验方法及结果见表2和3。
表1挥发油提取水平因素安排
检测指标:由于野菊花中蒙花苷具有抗菌、保肝作用,因此检测指标以蒙花苷提取率作为考察指标,根据野菊花药材质量标准规定,野菊花含蒙花苷不得少0.80%,本品实测没食子酸含量为1.81%;同时已挥发油提取率为指标,共同确定挥发油提取工艺。
挥发油提取率的样品制备
取相应比例桂枝饮片、海风藤饮片、野菊花饮片,加水,提取挥发油,记录挥发油体积;提取挥发油后的水提液滤过,记录水提液体积。
计算挥发油提取率
蒙花苷提取率的样品制备及测定
色谱柱aglientextend-c18(4.6mm×250mm,5μm),流动相甲醇:水:冰醋酸(26:23:1),柱温30℃,流速1ml/min,检测波长334nm。
对照品溶液的制备精密称取蒙花苷对照品适量,加甲醇制成每1ml含25μg的溶液,即得。
供试品溶液的制备分别量取正交试验制备所得水提液滤液重量约90g,蒸干,加甲醇适量使溶解,移至100ml量瓶中,超声处理(功率300w,频率50khz)30分钟,放冷,加甲醇稀释至刻度,摇匀,滤过;精密吸取续滤液1ml至25ml量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液、供试品溶液10μl,注入液相色谱仪,测定,计算,即得。
表2正交挥发油提取率结果分析
正交试验挥发油提取率结果分析:
表3正交试验蒙花苷提取率结果分析
根据数理统计理论,我们对表2和3的数据进行计算和分析。
由挥发油提取率的r值可知,各因素对挥发油提取影响顺序为:a>c>b,其中较主要的因素是a,其次是c、b;结合f值,以挥发油提取率为指标,推导其最佳因素水平组合为a2b2c2。由蒙花苷提取率的r′值可知,各因素对挥发油提取影响顺序为:a>b>c,其中较主要的因素是a,其次是b、c;结合f′值,从节约能源、缩短时间、节约成本考虑,以蒙花苷提取率为指标,推导其最佳因素水平组合为a2b2c2。即取桂枝、海风藤、野菊花药材,粉碎成最粗粉,加8倍量水提取挥发油4小时,收集挥发油;提取挥发油后的水提液滤过。
实施例2研磨法包合挥发油工艺的考察
本项目采用正交设计对工艺条件进行了优化,以挥发油的包合率为指标,考察了水与β-环糊精的比例、挥发油与β-环糊精的比例、研磨时间等工艺参数对挥发油包合的影响,用正交表l9(33)进行试验,方案见表4,实验方法及结果见表5。
表4挥发油包合水平因素安排
空白回收率的测定
精密量取挥发油5ml,平行2份,置1000ml圆底烧瓶中,均加水500ml,振摇混合,连接挥发油测定器与回流冷凝管。自冷凝管上端加水使充满挥发油测定器的刻度部分,并溢流入烧瓶为止。置电热套中缓慢加热至沸,并保持微沸5小时,至测定器中油量不再增加,停止加热,放置片刻,开启测定器下端活塞,将水缓慢放出,至油层上端到达刻度0线上面5mm处为止。放置1小时以上,再开启活塞使油层下降至其上端恰好与刻度0平齐,读取挥发油量,并计算空白回收率(%)
表5空白回收率数据
包合率的测定[0089]照《中国药典》2010版一部附录xd“挥发油测定法(甲法)”,取各试验所得β-环糊精包合物,置圆底烧瓶中,加入500ml纯净水,振摇混合,连接挥发油测定器与回流冷凝管。自冷凝管上端加水使充满挥发油测定器的刻度部分,并溢流入烧瓶为止。置电热套中缓慢加热至沸,并保持微沸5小时,至测定器中油量不再增加,停止加热,放置片刻,开启测定器下端活塞,将水缓慢放出,至油层上端到达刻度0线上面5mm处为止。放置1小时以上,再开启活塞使油层下降至其上端恰好与刻度0平齐,读取挥发油量。按下列公式计算包合物的包合率(%)。
表6挥发油包合正交实验结果分析
由包合率的r值可知,各因素对研磨法包合挥发油影响大小的顺序为:a>c>b,其中较主要的因素是a,其次是c、b。结合f值,以挥发油包合率为指标,推导其最佳因素水平组合为a2b2c2,即β-环糊精:水为1:3(w/v),挥发油:β-环糊精(v/w)为1:6,研磨包合25分钟。
实施例3醇提取工艺的考察
本项目采用正交设计对工艺条件进行了优化,分别考察了乙醇浓度(a)、乙醇用量(b)、提取时间(c)及提取次数(d)等因素对黄芪甲苷提取率的影响,用正交表l9(34)进行试验,方案见表7,实验方法及结果见表8。
表7醇提取水平因素安排
检测指标:由于黄芪药材为君药,黄芪含黄芪甲苷,因此,检测指标以黄芪甲苷提取率作为考察指标,根据黄芪药材质量标准规定,黄芪含黄芪甲苷不得少于0.05%,本品实测黄芪甲苷含量为0.10%。
醇提取的样品制备及测定
本实验中,每组1个处方量的药材,按表7方案提取,将收集的煎煮液分别称量取重量,备用。
黄芪甲苷测定:照高效液相色谱法(中国药典2000年版一部附录ⅵd)测定
色谱柱xb-c18(4.6mm×250mm,5μm),流动相乙腈:水(35:65),柱温30℃,漂移管温度68℃,流速1ml/min,气体流速2.0l/min。
对照品溶液的制备精密称取黄芪甲苷对照品适量,加甲醇制成每1ml含1mg的溶液,即得。
供试品溶液的制备取各正交试验乙醇提取液重量的1/10,蒸干,残渣加水10ml使溶解,用水饱和的正丁醇振摇提取4次,每次40ml,合并正丁醇液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液5μl、10μl与供试品溶液各5~10μl,注入液相色谱仪,测定,计算,即得。
表8实验结果及数据分析
由黄芪甲苷提取率的r值可知,各因素对其影响大小的顺序为:c>d>a>b,其中较主要的因素是c,其次是a、d、b,推导其最佳因素水平组合为a2b2c2d3。结合f值,乙醇提取2次和3次黄芪甲苷的提取率无显著差异,综合考虑得膏率情况、生产成本、节能降耗等因素,最终确定提取次数为2次,即黄芪、五加皮等乙醇提取工艺条件为:取黄芪、五加皮等药材,加70%乙醇回流提取两次,第一次加6倍量提取2小时,第二次加6倍量提取1.5小时,滤过,合并滤液,即得。
实施例4干燥方法的优选及得膏率的考察
本发明中稠膏的干燥方法通常采用常温常压干燥或真空干燥,为此我们取1个处方(2500g)药材共4份,分别黄芪、五加皮、浙贝母、刘寄奴、秦皮加70%乙醇回流提取两次,第一次加8倍量提取2小时,第二次加8倍量提取1小时,滤过,合并滤液,得乙醇提取液,备用;将醇沉上清液、乙醇提取液合并,减压回收乙醇,浓缩至相对密度1.20~1.25(60℃)的稠膏,分别采用常温常压干燥(常压,60~80℃)或真空干燥(-0.06~0.08mpa,60~80℃),结果见表9。
表9不同干燥方式所耗时间及样品得膏率
表9结果显示,当稠膏浓缩至1.20~1.24(60℃)时采用常压干燥耗时长,约为真空干燥2倍时间,而真空干燥由于样品在负压下形成了蜂窝状,使干燥物表面积增大,蒸发的水分在负压下迅速散发,因此干燥时间短,且干燥物疏松,易于粉碎,故宜采用真空干燥但真空干燥时稠膏也不宜太稀,太稀干燥时间延长,同时在负压下浸膏易溢出而损耗;真空干燥开始时负压不宜太大,温度不宜太高,否则易溢出,而应逐渐提高温度和负压;因此,稠膏相对密度浓缩至1.20~1.25(60℃)。
实施例5成型实验
根据上述实验结果可知,本品日服用生药材量为45g,本品总干膏得率为13%,计算得日服用总浸膏量为7.36g。片剂生产操作简单,具有使用方便、产品稳定性好、药物含量差异较小、贮存期长等优点,有利于大批量生产和贮运。其中微晶纤维素、可溶性淀粉、糊精、羧甲基淀粉钠作为颗粒成型的主要辅料,因此,我们取上述辅料与浸膏粉按表10进行实验,并分别以颗粒的流动性(休止角—漏斗法)、片剂的硬度与崩解时间为指标,考察处方的优劣。
表10片剂处方筛选结果
上述实验结果显示,以处方2、处方3的压片效果较佳,而处方2中崩解时限明显缩短,处方3崩解时限也较短,但颗粒得率较低,因此拟采用处方2,即干膏粉与糊精混匀,以乙醇制粒、干燥,加部分交1%硬脂酸镁,混匀,压片。
实施例6
黄芪750g桂枝250g秦皮500g五加皮500g野菊花500g海风藤500g刘寄奴500g浙贝母250g
β-环糊精2.30g糊精90g硬脂酸镁5.30g薄膜包衣粉16g
具体的制备方法包含如下步骤:
(1)水提挥发油:取桂枝250g、海风藤500g、野菊花500g,粉碎成最粗粉,加12倍量水提取2小时,收集挥发油;
(2)挥发油包合:用β-环糊精2.30g在20℃下研磨包合30min,过滤,干燥,得挥发油包合物;
(3)水提液浓缩与醇沉:提取挥发油后的水提液滤过,减压浓缩至相对密度1.10~1.15(60℃)的稠膏,放置至室温,边加入95%乙醇边搅拌使含醇量达55%,继续搅拌10min,室温静置24h,取上清液,滤过,即得醇沉上清液,备用;
(4)醇提:黄芪750g、五加皮500g、浙贝母250g、刘寄奴500g、秦皮500g,加65%乙醇回流提取两次,第一次加6倍量提取1.5小时,第二次加6倍量提取1小时,滤过,合并滤液,得乙醇提取液,备用;
(5)回收乙醇与浓缩:将醇沉上清液、乙醇提取液合并,减压回收乙醇,浓缩至相对密度1.20~1.25(60℃)的稠膏,备用;
(6)干燥、粉碎:取上述浸膏,置真空干燥器内干燥,粉碎,过80目筛,得提取干浸膏粉,置不锈钢桶内,备用;
(7)制粒与干燥:取上述干浸膏粉加糊精90g,混匀,以85%乙醇为粘合剂,湿法制粒,干燥,整粒,备用;
(8)压片:取上述干燥好的颗粒,再加硬脂酸镁5.30g、挥发油包合物,混匀,制成本品1000片(20150101批);
(9)薄膜包衣:取包薄膜衣粉16g加65%乙醇配置成混悬溶液,进行薄膜包衣,即得。
其中步骤(3)中滤液减压浓缩的压强是-0.06mpa~0.08mpa。
其中步骤(5)中药液减压的压强是-0.06mpa~-0.08mpa。
其中步骤(6)中置真空干燥器内干燥粉碎的压强为-0.08~0.09mpa,干燥温度为60℃。
其中步骤(6)中的混匀是在混合机中混合16分钟,测定混合物料的水份小于3.5%,压片控制硬度>7kg。
6.【检查】应符合片剂项下有关的各项规定(附录id)。
7.【薄层鉴别】
(1)将按原料比例制备好的芪桂痛风片,取8片,除去包衣,研细,加甲醇40ml,超声处理40min,滤过,滤液蒸干,残渣加水40ml使溶解,用水饱和的正丁醇萃取2次,每次60ml,合并正丁醇液,用1%氢氧化钠溶液洗涤2次,每次30ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺黄芪阴性对照品溶液;取黄芪对照药材1g,按供试品溶液的制备方法制成对照药材溶液;取黄芪甲苷对照品,加甲醇制成每1ml含0.8mg的黄芪甲苷对照品溶液;照薄层色谱法(2010年版药典附录ⅵb),分别吸取上述4种溶液各10μl,点于同一硅胶g薄层板上,以氯仿:甲醇:水(10:4:0.4)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,分别在日光下检视;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(2)将按原料比例制备好的芪桂痛风片,取6片,除去包衣,研细,加甲醇15ml,加热回流8min,放冷,滤过,滤液作为供试品溶液;按原料比例的制备方法,制成缺秦皮阴性对照品溶液;取秦皮对照药材1g,按供试品溶液的制备方法制成秦皮对照药材溶液;取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含1.5mg的混合溶液,作为对照品溶液;照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各6μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-甲酸(4:0.5:0.1)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(3)将按原料比例制备好的芪桂痛风片,取8片,除去包衣,研细,加浓氨试液3ml与二氯甲烷45ml,加热回流0.5h,放冷,滤过,滤液用0.1mol/l的盐酸溶液振摇提取1次,每次30ml,合并提取液,用浓氨调节ph到9用二氯甲烷30ml振摇提取1次,取二氯甲烷滤液,蒸干,加无水乙醇1ml溶解,作为供试品溶液;按原料比例的制备方法,制成缺浙贝母阴性对照品溶液;取浙贝母对照药材3g,按供试品溶液的制备方法制成浙贝母对照药材溶液;另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含1.5mg的混合溶液,作为对照品溶液;照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-甲醇-浓氨试液(13:0.8:0.4)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点。
(4)将按原料比例制备好的芪桂痛风片,取5片,除去包衣,研细,加二氯甲烷55ml,超声处理40min,滤过,滤液蒸干,残渣加二氯甲烷1ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺五加皮阴性对照品溶液;取五加皮对照药材0.5g,按供试品溶液的制备方法制成五加皮对照药材溶液;照薄层色谱法(2015年版《中国药典》附录ⅵb)试验,吸取上述两种溶液各5μl,分别点于同一硅胶g薄层板上,以石油醚(60~90℃)-丙酮-异丙醇-甲酸(6:1:0.2:0.04)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,在日光下检视;供试品、对照药材在薄层色谱图相同位置上,显相同颜色的斑点;
(5)将按原料比例制备好的芪桂痛风片,取6片,除去包衣,研细,加二氯甲烷35ml,超声40min,滤过,加入等体积1%naoh溶液萃取4次,合并萃取液,加盐酸酸化至ph值为2~4,再用等体积二氯甲烷萃取4次,合并萃取液,挥去二氯甲烷,残渣加甲醇0.5ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺桂枝阴性对照品溶液;取桂枝对照药材1g,按供试品溶液的制备方法制成桂枝对照药材溶液;取肉桂酸对照品,加乙醇制成每1ml含0.8mg的溶液,作为对照品溶液;照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各10μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(3:3:0.02)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点;
8.含量测定:照高效液相色谱法(中国药典2010年版一部附录ⅵd)测定,具体按照以下步骤a~步骤d进行:
a、色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;流动相:乙腈-水(35:65)为流动相;流速:1ml·min-1;柱温:30℃;气体流速:1.5l/min;漂移管温度:105℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
b、溶液制备:
供试品溶液的制备:取重量差异项下的本品10片,除去包衣,研细,称取4.5g,精密称定,精密称定,加甲醇45ml,超声提取40min,滤过,回收溶剂并浓缩至干,残渣加水12ml,微热使溶解,用水饱和的正丁醇振摇提取4次,每次45ml,合并正丁醇提取液,用氨试液充分洗涤3次,每次45ml,弃去氨液,正丁醇液蒸干,残渣加水15ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水50ml洗脱,弃去水液,再用40%乙醇40ml洗脱,弃去洗脱液,继用85%乙醇90ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,即得。
黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品适量,加甲醇分别制成每1ml含0.30mg、每1ml含0.60mg的对照品溶液,即得。
c、专属性试验:按照步骤a所述的色谱条件,分别精密吸取供试品溶液、黄芪阴性对照溶液、对照品溶液各8μl,注入高效液相色谱仪,测定,以外标两点法对数方程计算,即得。
d、测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。
8.每片含黄芪以黄芪甲苷(c41h68o14)计,不少于0.16mg。
实施例7
黄芪750g桂枝250g秦皮500g五加皮500g野菊花500g海风藤500g刘寄奴500g浙贝母250g
β-环糊精2.70g糊精110g硬脂酸镁5.60g薄膜包衣粉18g
具体的制备方法包含如下步骤:
(1)水提挥发油:取桂枝250g、海风藤500g、野菊花500g,粉碎成最粗粉,加10倍量水提取4小时,收集挥发油;
(2)挥发油包合:用β-环糊精2.70g在28℃下研磨包合24min,过滤,干燥,得挥发油包合物;
(3)水提液浓缩与醇沉:提取挥发油后的水提液滤过,减压浓缩至相对密度1.10~1.15(60℃)的稠膏,放置至室温,边加入95%乙醇边搅拌使含醇量达70%,继续搅拌20min,室温静置16h,取上清液,滤过,即得醇沉上清液,备用;
(4)醇提:黄芪750g、五加皮500g、浙贝母250g、刘寄奴500g、秦皮500g,加70%乙醇回流提取两次,第一次加8倍量提取2.0小时,第二次加8倍量提取1.5小时,滤过,合并滤液,得乙醇提取液,备用;
(5)回收乙醇与浓缩:将醇沉上清液、乙醇提取液合并,减压回收乙醇,浓缩至相对密度1.20~1.25(60℃)的稠膏,备用;
(6)干燥、粉碎:取上述浸膏,置真空干燥器内干燥,粉碎,过80目筛,得提取干浸膏粉,置不锈钢桶内,备用;
(7)制粒与干燥:取上述干浸膏粉加糊精110g,混匀,以90%乙醇为粘合剂,湿法制粒,干燥,整粒,备用;
(8)压片:取上述干燥好的颗粒,再加硬脂酸镁5.60g、挥发油包合物,混匀,制成本品1000片(20150102批);
(9)薄膜包衣:取包薄膜衣粉18g加75%乙醇配置成混悬溶液,进行薄膜包衣,即得。
其中步骤(3)中滤液减压浓缩的压强是-0.06mpa~0.08mpa。
其中步骤(5)中药液减压的压强是-0.06mpa~-0.08mpa。
其中步骤(6)中置真空干燥器内干燥粉碎的压强为-0.08~0.09mpa,干燥温度为60~75℃。
其中步骤(6)中的混匀是在混合机中混合20分钟,测定混合物料的水份小于3.5%,压片控制硬度>7kg。
6.【检查】应符合片剂项下有关的各项规定(附录id)。
7.【薄层鉴别】
(1)取本品8片,除去包衣,研细,加甲醇50ml,超声处理35min,滤过,滤液蒸干,残渣加水30ml使溶解,用水饱和的正丁醇萃取3次,每次40ml,合并正丁醇液,用1%氢氧化钠溶液洗涤3次,每次25ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取黄芪对照药材2g,同法制成对照药材溶液。另取黄芪甲苷对照品,加甲醇制成每1ml含1.0mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录vib)试验,吸取上述三种溶液各8μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-水(12:6.5:1.5)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(2)取本品6片,除去包衣,研细,加甲醇25ml,加热回流10min,放冷,滤过,滤液作为供试品溶液。另取秦皮对照药材2.0g,同法制成对照药材溶液。另取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录vib)试验,吸取上述三种溶液各6μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-甲酸(5:0.8:0.3)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。
(3)取本品9片,除去包衣,研细,加浓氨试液4ml与二氯甲烷52ml,加热回流1h,放冷,滤过,滤液用0.15mol/l的盐酸溶液振摇提取2次,每次20ml,合并提取液,用浓氨调节ph到10,用二氯甲烷振摇提取2次,每次25ml,合并三氯甲烷液,蒸干,加无水乙醇1ml溶解,作为供试品溶液。另取浙贝母对照药材4g,同法制成对照药材溶液。另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录vib)试验,吸取上述三种溶液各6μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-甲醇-浓氨试液(16:1.5:0.6)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点。
(4)取本品8片,除去包衣,研细,加二氯甲烷65ml,超声处理30min,滤过,滤液蒸干,残渣加二氯甲烷2ml使溶解,作为供试品溶液。另取五加皮对照药材1g,同法制成对照药材溶液。照薄层色谱法(2015年版《中国药典》附录vib)试验,吸取上述两种溶液各8μl,分别点于同一硅胶g薄层板上,以石油醚(60~90℃)-丙酮-异丙醇-甲酸(10:1.5:0.3:0.06)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,在日光下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点。
(5)取本品8片,除去包衣,研细,加二氯甲烷45ml,超声30min,滤过,加入等体积1%naoh溶液萃取2次,合并萃取液,加盐酸酸化至ph值为3~4,再用等体积二氯甲烷萃取2次,合并萃取液,挥去二氯甲烷,残渣加甲醇1ml使溶解,作为供试品溶液。另取桂枝对照药材2g,同制成对照药材溶液。另取肉桂酸对照品,加乙醇制成每1ml含1.2mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(4:4:0.08)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点。
8.含量测定:照高效液相色谱法(中国药典2010年版一部附录ⅵd)测定,具体按照以下步骤a~步骤d进行:
a、色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;流动相:乙腈-水(30:70)为流动相;流速:1ml·min-1;柱温:30℃;气体流速:2.0l/min;漂移管温度:80℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
b、溶液制备:
供试品溶液的制备:取重量差异项下的本品10片,除去包衣,研细,称取4.0g,精密称定,加甲醇40ml,超声提取30min,滤过,回收溶剂并浓缩至干,残渣加水10ml,微热使溶解,用水饱和的正丁醇振摇提取3次,每次40ml,合并正丁醇提取液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,残渣加水12ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水45ml洗脱,弃去水液,再用35%乙醇30ml洗脱,弃去洗脱液,继用78%乙醇80ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,即得。
黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品适量,加甲醇分别制成每1ml含0.28mg、每1ml含0.54mg的对照品溶液,即得。
c、专属性试验:按照步骤a所述的色谱条件,分别精密吸取供试品溶液、黄芪阴性对照溶液、对照品溶液各8μl,注入高效液相色谱仪,测定,以外标两点法对数方程计算,即得。
d、测定法分别精密吸取对照品溶液与供试品溶液各8μl,注入液相色谱仪,测定,即得。
8.每片含黄芪以黄芪甲苷(c41h68o14)计,不少于0.16mg。
实施例8
黄芪750g桂枝250g秦皮500g五加皮500g野菊花500g海风藤500g刘寄奴500g浙贝母250g
β-环糊精3.10g糊精120g硬脂酸镁6.00g薄膜包衣粉20g
具体的制备方法包含如下步骤:
(1)水提挥发油:取桂枝250g、海风藤500g、野菊花500g,粉碎成最粗粉,加12倍量水提取2小时,收集挥发油;
(2)挥发油包合:用β-环糊精3.10g在35℃下研磨包合15min,过滤,干燥,得挥发油包合物;
(3)水提液浓缩与醇沉:提取挥发油后的水提液滤过,减压浓缩至相对密度1.10~1.15(60℃)的稠膏,放置至室温,边加入95%乙醇边搅拌使含醇量达85%,继续搅拌30min,室温静置8h,取上清液,滤过,即得醇沉上清液,备用;
(4)醇提:黄芪750g、五加皮500g、浙贝母250g、刘寄奴500g、秦皮500g,加75%乙醇回流提取两次,第一次加12倍量提取2.5小时,第二次加12倍量提2小时,滤过,合并滤液,得乙醇提取液,备用;
(5)回收乙醇与浓缩:将醇沉上清液、乙醇提取液合并,减压回收乙醇,浓缩至相对密度1.20~1.25(60℃)的稠膏,备用;
(6)干燥、粉碎:取上述浸膏,置真空干燥器内干燥,粉碎,过100目筛,得提取干浸膏粉,置不锈钢桶内,备用;
(7)制粒与干燥:取上述干浸膏粉加糊精120g,混匀,以95%乙醇为粘合剂,湿法制粒,干燥,整粒,备用;
(8)压片:取上述干燥好的颗粒,再加硬脂酸镁6.00g、挥发油包合物,混匀,制成本品1000片(20150201批);
(9)薄膜包衣:取包薄膜衣粉20g加85%乙醇配置成混悬溶液,进行薄膜包衣,即得。
其中步骤(3)中滤液减压浓缩的压强是-0.06mpa~0.08mpa。
其中步骤(5)中药液减压的压强是-0.06mpa~-0.08mpa。
其中步骤(6)中置真空干燥器内干燥粉碎的压强为-0.08~0.09mpa,干燥温度为60~75℃。
其中步骤(6)中的混匀是在混合机中混合20分钟,测定混合物料的水份小于3.5%,压片控制硬度>7kg。
6.【检查】应符合片剂项下有关的各项规定(附录id)。
7.【薄层鉴别】
(1)将按原料比例制备好的芪桂痛风片,取10片,除去包衣,研细,加甲醇60ml,超声处理25min,滤过,滤液蒸干,残渣加水40ml使溶解,用水饱和的正丁醇萃取4次,每次40ml,合并正丁醇液,用1%氢氧化钠溶液洗涤4,每次15ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇3ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺黄芪阴性对照品溶液;取黄芪对照药材3g,按供试品溶液的制备方法制成对照药材溶液;取黄芪甲苷对照品,加甲醇制成每1ml含1.5mg的黄芪甲苷对照品溶液;照薄层色谱法(2010年版药典附录ⅵb),分别吸取上述4种溶液各4μl,点于同一硅胶g薄层板上,以氯仿:甲醇:水(13:7:2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰,分别在日光下检视;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(2)将按原料比例制备好的芪桂痛风片,取9片,除去包衣,研细,加甲醇30ml,加热回流8min,放冷,滤过,滤液作为供试品溶液;按原料比例的制备方法,制成缺秦皮阴性对照品溶液;取秦皮对照药材3g,按供试品溶液的制备方法制成秦皮对照药材溶液;取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含2.5mg的混合溶液,作为对照品溶液;照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各1μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-甲酸(6:1:0.5)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点;
(3)将按原料比例制备好的芪桂痛风片,取12片,除去包衣,研细,加浓氨试液6ml与二氯甲烷60ml,加热回流0.5h,放冷,滤过,滤液用0.2mol/l的盐酸溶液振摇提取3次,每次30ml,合并提取液,用浓氨调节ph到9~11用二氯甲烷振摇提取3次,每次10ml,合并二氯甲烷液,蒸干,加无水乙醇1ml溶解,作为供试品溶液;按原料比例的制备方法,制成缺浙贝母阴性对照品溶液;取浙贝母对照药材6g,按供试品溶液的制备方法制成浙贝母对照药材溶液;另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含2mg的混合溶液,作为对照品溶液;照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各3μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-甲醇-浓氨试液(17:2:1)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液;供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的斑点;
(4)将按原料比例制备好的芪桂痛风片,取8片,除去包衣,研细,加二氯甲烷70ml,超声处理25min,滤过,滤液蒸干,残渣加二氯甲烷3ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺五加皮阴性对照品溶液;取五加皮对照药材2g,按供试品溶液的制备方法制成五加皮对照药材溶液;照薄层色谱法(2015年版《中国药典》附录ⅵb)试验,吸取上述两种溶液各1μl,分别点于同一硅胶g薄层板上,以石油醚(60~90℃)-丙酮-异丙醇-甲酸(12:2:0.5:0.1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,在日光下检视;供试品、对照药材在薄层色谱图相同位置上,显相同颜色的斑点;
(5)将按原料比例制备好的芪桂痛风片,取10片,除去包衣,研细,加二氯甲烷50ml,超声25min,滤过,加入等体积1%naoh溶液萃取4次,合并萃取液,加盐酸酸化至ph值为2~4,再用等体积二氯甲烷萃取4次,合并萃取液,挥去二氯甲烷,残渣加甲醇0.5ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺桂枝阴性对照品溶液;取桂枝对照药材3g,按供试品溶液的制备方法制成桂枝对照药材溶液;取肉桂酸对照品,加乙醇制成每1ml含1.5mg的溶液,作为对照品溶液;照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各5μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(5:5:0.1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视;供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点;
8.【检查】应符合片剂项下有关的各项规定(附录id)。
7.含量测定:照高效液相色谱法(中国药典2010年版一部附录ⅵd)测定,具体按照以下步骤a~步骤d进行:
a、色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;流动相:乙腈-水(35:75)为流动相;流速:1ml·min-1;柱温:30℃;气体流速:1.5l/min;漂移管温度:105℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
b、溶液制备:
供试品溶液的制备:取重量差异项下的本品10片,除去包衣,研细,称取4.5g,精密称定,加甲醇45ml,超声提取25min,滤过,回收溶剂并浓缩至干,残渣加水12ml,微热使溶解,用水饱和的正丁醇振摇提取4次,每次45ml,合并正丁醇提取液,用氨试液充分洗涤3次,每次45ml,弃去氨液,正丁醇液蒸干,残渣加水15ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水50ml洗脱,弃去水液,再用40%乙醇40ml洗脱,弃去洗脱液,继用85%乙醇70ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,即得。
黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品适量,加甲醇分别制成每1ml含0.30mg、每1ml含0.60mg的对照品溶液,即得。
c、专属性试验:按照步骤a所述的色谱条件,分别精密吸取供试品溶液、黄芪阴性对照溶液、对照品溶液各10μl,注入高效液相色谱仪,测定,以外标两点法对数方程计算,即得。
d、测定法分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,测定,即得。
8.每片含黄芪以黄芪甲苷(c41h68o14)计,不少于0.16mg。
实验例9:本发明中各药味鉴别方法的筛选
(1)黄芪药材的薄层鉴别
方法一、取本发明提供的芪桂痛风片6片,除去包衣,研细,加乙醇50ml,超声处理25min,滤过,滤液蒸干,残渣加水10ml使溶解,滤过,滤液加正丁醇提取2次,每次40ml(轻轻振摇),合并正丁醇提取液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。按原料比例同法制成缺黄芪阴性对照品溶液。另取黄芪对照药材3g,同法制成对照药材溶液。另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水(13:6:1)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。结果显示,供试品、对照药材与对照品在薄层色谱图相同位置上,均未显现相同的斑点。置紫外光灯(365nm)下检视,在薄层色谱图相同位置上无相同颜色的荧光斑点,方法专属性差。
方法二、取本发明提供的芪桂痛风片6片,除去包衣,研细,加甲醇40ml,回流提取35min,滤过,滤液蒸干,残渣加水30ml使溶解,用水饱和的正丁醇萃取2次,每次40ml,合并正丁醇液,用1%氢氧化钠溶液洗涤2次,每次20ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。按原料比例同法制成缺黄芪阴性对照品溶液。另取黄芪对照药材3g,同法制成对照药材溶液。另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-水(14:7:2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,但样品斑点较浅。
方法三、取本发明提供的芪桂痛风片6片,除去包衣,研细,加甲醇40ml,超声处理20min,滤过,滤液蒸干,残渣加水30ml使溶解,用水饱和的正丁醇萃取2次,每次40ml,合并正丁醇液,用1%氢氧化钠溶液洗涤2次,每次20ml,再用以正丁醇饱和的水洗至中性,正丁醇液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。按原料比例同法制成缺黄芪阴性对照品溶液。另取黄芪对照药材3g,同法制成对照药材溶液。另取黄芪甲苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-水(13:6:2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。
结论:综上所述,优选方法三作为本制剂中黄芪的质量控制方法之一。
(2)秦皮药材的薄层鉴别
方法一、取本发明提供的芪桂痛风片6片,除去包衣,研细,加甲醇20ml,加热回流10min,放冷,滤过,滤液作为供试品溶液。按原料比例同法制成缺秦皮阴性对照品溶液。另取秦皮对照药材2g,同法制成对照药材溶液。另取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各4μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-甲酸(6:1:0.5)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。
方法二、取本发明提供的芪桂痛风片6片,除去包衣,研细,加甲醇20ml,加热回流10min,放冷,滤过,滤液置蒸发皿中,于水浴上挥干,残渣加入5ml甲醇溶解,作为供试品溶液。按原料比例同法制成缺秦皮阴性对照品溶液。另取秦皮对照药材2g,同法制成对照药材溶液。另取秦皮素、秦皮甲素、秦皮乙素对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各4μl,分别点于同一硅胶g薄层板上,以二氯甲烷-甲醇-甲酸(7:0.5:0.5)为展开剂,展开,取出,晾干,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,有相同颜色的荧光斑点,但主斑点的分离度较差。
结论:综上所述,优选方法一作为本制剂中秦皮的质量控制方法之一。
(3)浙贝母药材的薄层鉴别
取本发明提供的芪桂痛风片10片,除去包衣,研细,加浓氨试液4ml与二氯甲烷50ml,加热回流1h,放冷,滤过,滤液用0.15mol/l的盐酸溶液振摇提取2次,每次20ml,合并提取液,用浓氨调节ph到10,用二氯甲烷振摇提取2次,每次20ml,合并三氯甲烷液,蒸干,加无水乙醇1ml溶解,作为供试品溶液。按原料比例的制备方法,制成缺浙贝母阴性对照品溶液;另取浙贝母对照药材4g,同法制成对照药材溶液。另取贝母素甲对照品、贝母素乙对照品,加无水乙醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录ⅵb)试验,吸取上述三种溶液各6μl,分别点于同一硅胶g薄层板上,以乙酸乙酯-甲醇-浓氨试液(17:2:1)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液。结果显示,供试品、对照药材与对照品在薄层色谱图相同位置上,,显相同颜色的斑点。结论:该方法作为本制剂中浙贝母的质量控制。
(4)五加皮药材的薄层鉴别
方法一、取本发明提供的芪桂痛风片5片,除去包衣,研细,加乙醚20ml,超声处理10分钟,过滤,弃去乙醚液,重复3次。残渣挥干乙醚,加甲醇20ml,超声处理30分钟,滤过,滤液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液;按原料比例的制备方法,制成缺五加皮阴性对照品溶液;另取紫丁香苷对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录ⅵb)试验,吸取供试品溶液、五加皮阴性对照溶液、对照品溶液各5μl,分别点于同硅胶胶g薄层板上,以三氯甲烷-甲醇(5:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。结果表明,制剂及阴性制剂在与对照品色谱相同的位置上因其他成分干扰,无相同颜色的斑点。
方法二、取本发明提供的芪桂痛风片10片,除去包衣,研细,加乙醇100ml,超声处理30min,滤过,回收乙醇至无醇味,加入2倍体积的5%nahco3水溶液,用乙酸乙酯振摇提取3次,每次25ml。弃去乙酸乙酯液(除掉中性或酚类成分),5%nahco3水溶液层加4%的盐酸调ph=1-2,加入乙醚萃取,萃取三次,乙醚萃取液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液;取五加皮药材2g,同法制成五加皮药材溶液;另取五加皮阴性制剂,同法制得阴性对照溶液;再取原儿茶酸对照品,加甲醇制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法(2010年版药典附录ⅵb)试验,吸取供试品溶液、五加皮阴性对照溶液各2μl,原儿茶酸对照品溶液1μl,五加皮药材溶液8μl,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲酸(5:3:0.1)为展开剂,展开,取出,晾干,喷以2%三氯化铁溶液-1%铁氰化钾溶液(1:1)(临用配制),放置至斑点显色清晰。结果显示,供试品、阴性制剂、对照药材与对照品在薄层色谱图相同位置上,均显相同颜色的斑点,阴性干扰严重,专属性差。
方法三、取本发明提供的芪桂痛风片6片,除去包衣,研细,加二氯甲烷45ml,超声处理20min,滤过,滤液蒸干,残渣加二氯甲烷2ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺五加皮阴性对照品溶液;另取五加皮对照药材1g,同法制成对照药材溶液。照薄层色谱法(2015年版《中国药典》附录ⅵb)试验,吸取上述两种溶液各5μl,分别点于同一硅胶g薄层板上,以石油醚(60~90℃)-丙酮-异丙醇-甲酸(12:2:0.5:0.1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,在日光下检视。结果显示,供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点。
结论:综上所述,优选方法三作为本制剂中五加皮的质量控制方法之一。
(5)桂枝药材的薄层鉴别
方法一、取本发明提供的芪桂痛风片6片,除去包衣,研细,加乙醚30ml,冷浸12h,滤过,滤液用无水硫酸钠1.5g脱水,滤过,加入40ml1%naoh溶液萃取,取碱萃取层加2%盐酸酸化(ph值5-6),再用乙醚萃取(20ml/次,2次),挥去乙醚,残渣加甲醇0.5ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺桂枝阴性对照品溶液;另取桂枝对照药材2g,同制成对照药材溶液。另取肉桂酸对照品,加乙醇制成每1ml含1.5mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各10μl,分别点于同一硅胶gf254薄层板上,以石油醚(30~60℃)-甲酸乙酯-甲酸(15︰4︰1)上层溶液为为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。结果显示,供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点,但供试品斑点显色不清晰。
方法二、取本发明提供的芪桂痛风片8片,除去包衣,研细,加二氯甲烷45ml,冷浸10小时,滤过,加入1%naoh溶液20ml萃取,萃取3次,合并萃取液,加稀盐酸酸化至ph值为5-6,再用二氯甲烷萃取(20ml/次,4次),挥去二氯甲烷,残渣加甲醇1ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺桂枝阴性对照品溶液;另取桂枝对照药材2g,同制成对照药材溶液。另取肉桂酸对照品,加乙醇制成每1ml含1.5mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(6:5:0.1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。结果显示,供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点,无阴性干扰,专属性较好,但供试品制备时间较长。
方法三、取本发明提供的芪桂痛风片8片,除去包衣,研细,加二氯甲烷45ml,超声15min,滤过,加入等体积2%naoh溶液萃取4次,合并萃取液,加盐酸酸化至ph值为3~4,再用等体积二氯甲烷萃取4次,合并萃取液,挥去二氯甲烷,残渣加甲醇1ml使溶解,作为供试品溶液。按原料比例的制备方法,制成缺桂枝阴性对照品溶液;另取桂枝对照药材2g,同制成对照药材溶液。另取肉桂酸对照品,加乙醇制成每1ml含1.5mg的溶液,作为对照品溶液。照薄层色谱法(2010年版《中国药典》附录ⅵb)试验,吸取上述三种溶液各8μl,分别点于同一硅胶gf254薄层板上,以正己烷-乙醚-冰醋酸(6:5:0.1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。结果显示,供试品、对照药材与对照品在薄层色谱图相同位置上,显相同颜色的斑点。
结论:综上所述,优选方法三作为本制剂中桂枝的质量控制方法之一。
实验例10:黄芪甲苷的含量测定
1、色谱条件的选择:对芪桂痛风片中黄芪甲苷进行测定,色谱柱:agilentzorbaxextendc18(4.6mm×250mm,5μm);流动相:乙腈-水(35:65)为流动相;流速:1.0ml·min-1;柱温:30℃;气体流速:1.5l/min;漂移管温度:55℃;理论塔板数按黄芪甲苷峰计算应不低于4000;
2、溶液制备
2.1、供试品溶液的制备:取重量差异项下的本发明提供的芪桂痛风片8片,除去包衣,研细,称取4g,精密称定,加甲醇35~45ml,超声提取20~40min,滤过,回收溶剂并浓缩至干,残渣加水8ml,微热使溶解,用水饱和的正丁醇振摇提取2次,每次35ml,合并正丁醇提取液,用氨试液充分洗涤2次,每次35ml,弃去氨液,正丁醇液蒸干,残渣加水4ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水40ml洗脱,弃去水液,再用35%乙醇30ml洗脱,弃去洗脱液,继用70%乙醇70ml洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,即得。
2.2、黄芪甲苷对照品溶液的制备:取黄芪甲苷对照品适量,加甲醇分别制成每1ml含0.26mg、每1ml含0.52mg的对照品溶液,即得。
2.3、阴性样品溶液的制备:按原料比例和工艺,制成不含当归药材的阴性样品,按供试品溶液的制备方法制备阴性对照溶液。
3、专属性试验:
按照步骤1所述的色谱条件,分别精密吸取供试品溶液、黄芪阴性对照溶液、对照品溶液各10μl,注入高效液相色谱仪,测定,以外标两点法对数方程计算,即得。黄芪甲苷色谱峰保留时间在10.4min左右,阴性样品溶液在相应位置无色谱峰。表明阴性样品无干扰,本法专属性强。
4、线性关系的考察精密称取黄芪甲苷24.95mg,置25ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得(每1ml含0.998mg的黄芪甲苷)。取对照品适量,分别加甲醇稀释成以下浓度:789.40μg/ml;479.04μg/ml;191.62μg/ml;76.65μg/ml;30.66μg/ml。分别吸取上述各浓度的对照品溶液10μl,注入高效液相色谱仪。以进样浓度(μg/ml)的对数为横坐标(x),峰面积的对数为纵坐标(y),进行回归计算,即得黄芪甲苷的线性回归方程为:y=1.4813x+0.2112,相关系数r=0.9996。实验结果表明:黄芪甲苷在30.66~998.00μg/ml浓度范围内呈良好线性。
表11黄芪甲苷线性关系
5、精密度试验精密吸取含黄芪甲苷对照品溶液,注入高效液相色谱仪,重复进样6次,每次10μl,测定峰面积,计算rsd为1.26%,表明精密度良好。
6、稳定性试验取重量差异项下的一批制剂适量,照[含量测定]“供试品溶液的制备”项下方法制备6份供试品溶液,分别于0、2、4、8、12、24小时注入液相色谱仪,计算黄芪甲苷含量,rsd值为1.92%。结果表明供试品溶液在24小时内稳定。
7、重复性试验取重量差异项下的一批制剂适量,平行6份,照[含量测定]“供试品溶液的制备”项下方法制备供试品溶液,精密吸取各供试品溶液10μl,测定峰面积,平均含量为0.8996mg/g,其rsd值为1.24%。结果表明重复性良好。
8、加样回收率试验取重量差异项下的一批制剂(适量,研细,称取2g,精密称定,平行9份,计算每份中黄芪甲苷的含量。上述样品每3份分为1组,每组分别按含黄芪甲苷量的0.8倍、1.0倍、1.2倍准确加入黄芪甲苷对照品,照[含量测定]“供试品溶液的制备”项下方法制备供试品溶液,精密吸取各供试品溶液10μl,注入高效液相色谱仪,测定峰面积,计算含量及其rsd值。平均回收率99.89%,rsd=2.14%,结果表明本法黄芪甲苷的加样回收率结果良好。
9、样品测定取本发明提供的芪桂痛风片适量,除去包衣,研细,称取5g,精密称定,置索氏提取器中,加甲醇,冷浸过夜,再加甲醇适量,加热回流2小时,提取液回收溶剂并浓缩至干,残渣加水,微热使溶解,用水饱和的正丁醇振摇提取4次,每次40ml,合并正丁醇提取液,用氨试液充分洗涤2次,每次40ml,弃去氨液,正丁醇液蒸干,残渣加水5ml使溶解,放冷,通过d101型大孔吸附树脂柱(内径为1.5cm,柱高为12cm),以水洗脱,弃去水液,再用35%乙醇洗脱,弃去洗脱液,继用75%乙醇洗脱,收集洗脱液,蒸干,残渣加甲醇溶解并转移至10ml量瓶中,加甲醇至刻度,摇匀,即得。精密吸取对照品溶液、供试品溶液各10μl,注入液相色谱仪,测定,以外标两点法对数方程计算,即得。
10、芪桂痛风片中黄芪甲苷含量见表12
表12芪桂痛风片中黄芪甲苷含量
根据表10测定的结果,平均每片含黄芪甲苷0.19mg,考虑药材来源、操作过程等因素的差异,因此,考虑本品每片约含0.75g黄芪药材,按照《中国药典》2010年版一部“黄芪”药材项下规定的“含黄芪甲苷不得少于0.040%”的最低限度,结合本品实际生产黄芪甲苷的平均转移率为60%,经计算,本品每片中含黄芪甲苷的最低含量应为0.16mg。根据上述试验结果,考虑到制剂中黄芪甲苷的含量限度建立应综合本品药材中黄芪甲苷的含量、提取转移率、制剂生产过程损耗等因素,并应积累多批样品含量测定的数据后最终制订本品中黄芪甲苷的含量限度。因此,暂定本品每片含黄芪以黄芪甲苷(c41h68o14)计,不得少于0.16mg。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!