连接子药物及包含其的抗体药物复合物的制作方法
本发明系有关于一种连接子药物(linker-drug)及包含其的抗体药物复合物(antibody-drug conjugate;ADC),特别系有关于一种具有高均质性的连接子药物及包含其的抗体药物复合物。
背景技术:
在癌症治疗方法中,许多细胞毒性药物分子因为无法选择性地毒杀癌细胞而无法用于癌症治疗。因此,已发展出抗体药物复合物(ADC)作为新型的癌症治疗药物。抗体药物复合物一般是由抗体、连接子、和药物三个部分所组成,其中抗体以连接子和药物连接。抗体药物复合物的作用机制是先通过抗体对于相应抗原的高度辨识能力来识别肿瘤细胞上专一或过度表现的抗原而与之结合,形成的结合物以内吞的方式进入肿瘤细胞时,连接的细胞毒性药物分子也同时被带入肿瘤细胞。在肿瘤细胞中,抗体被分解或是连接子断裂使得细胞毒性药物分子释放,进而杀死目标肿瘤细胞,达到选择性的毒杀作用。
ADC中使用的连接子需要满足几个要求:当在人血浆中循环时,连接子需要稳定以防止药物的早期释放;在内化到肿瘤细胞中时,可切割的连接子在一定条件下可被切割以释放药物,而对于不可切割的连接子,药物部分以包含药物、连接子、和衍生自蛋白酶降解配体的氨基酸残基的活性形式释放。
在目前使用的临床ADC结构中,药物通过不同的连接子连接至赖氨酸残基或铰链区半胱氨酸残基(在链间二硫键完全/部分还原的后)。最适化的DAR(药物抗体比)较佳为2~4。抗体表面大量的赖氨酸残基和非选择性接合模式导致接合位点和接合药物数量的不确定性。例如:在(ado-trastuzumab emtansine;Roche)中有几十种可能的接合位点,它们属于基于赖氨酸的接合。类似地,尽管抗体在铰链区附近含有四对可还原的链间二硫键,但必须部分还原和接合才能使ADC具有最适的平均DAR2~4。由于一般使用的还原剂(DTT,TCEP等)不能选择性还原铰链区二硫键,因此得到的接合产物也不均匀。例如,(本妥昔单抗(Brentuximab)vedotin;Seattle Genetics),其属于基于半胱氨酸的接合且包含具有DAR为0、2、4、6和8的的接合物。即使对于具有特定DAR值的部分,也是包含接合在不同接合位点的接合物的混合物。ADC产物的异质性可能最终导致不同组分的不同药物代谢动力学(pharmacokinetics,PK)、功效和毒性性质。此外,ADC产品的异质性也可能导致聚集和半衰期下降。
为了克服ADC产物高度异质性的问题,近年来,位点特异性(site-specific)接合技术成为控制接合位点和药物负载化学计量(stoichiometrics)的热点。然而,这些技术中的抗体或蛋白质大部分都经过基因工程。因为需要进行实质性的工作和特别的关注以筛选具有有利的突变位点的抗体,以进一步进行药物接合或聚乙二醇化,这样的诱变可能是耗时且不具有成本效益的。
另外,马来酰亚胺(maleimide)在大多数临床ADC中被用作抗体的接合位点。然而,马来酰亚胺很容易解离,导致半衰期短。在这种情况下,抗体和药物之间的连接体在ADC内化到肿瘤细胞之前被破坏,早期释放的药物不能特异性地杀死目标肿瘤细胞,并且不能实现选择性毒性作用。
因此,目前亟需一种具有高均质性(homogeneity)、高稳定性的抗体药物复合物以提高其药效和在血浆中的稳定度。
技术实现要素:
根据一实施例,本发明提供一种如化学式(I)所示的连接子药物。于化学式(I)中,C为接合连接子,L为连接子单元,D为药物单元,且m为从1至4的整数。接合连接子的结构如化学式(II)所示。于化学式(II)中,X为离去基,R1和R2同时为单键或–NH–,Z为芳基、经取代的芳基、杂芳基、经取代的杂芳基、直链烷基、环烷基、杂环烷基、或前述的组合。化学式(II)中的波浪线表示与L的共价连接位。
C-(L-D)m (I)
根据另一实施例,本发明也提供一种如化学式(IV)所示的抗体药物复合物(ADC)。于化学式(IV)中,A为全长抗体、抗体片段、蛋白质、或多肽;C’-(L-D)m为连接子药物,其中C’为接合连接子;L为连接子单元;D为药物单元;且m为从1至4的整数。其中,A通过分别存在于A的两个半胱氨酸残基中的两个巯基(thiol groups)与该连接子药物接合。A-C’包括如化学式(V)所示的结构。其中,R1和R2同时为单键或–NH–;Z为芳基、经取代的芳基、杂芳基、经取代的杂芳基、直链烷基、环烷基、杂环烷基、或前述的组合。其中,化学式(V)的波浪线表示与L的共价连接位;A’表示A的剩余部分,其通过分别存在于A的两个半胱氨酸残基中的两个巯基(thiol groups)与该连接子药物接合。
A-C’-(L-D)m (IV)
为让本发明的上述和其他目的、特征与优点能更明显易懂,下文特举出较佳实施例,并配合所附图式,作详细说明如下:
附图说明
图1为根据本发明一实施例显示赫赛汀(Herceptin)-连接子药物1的HIC图谱。
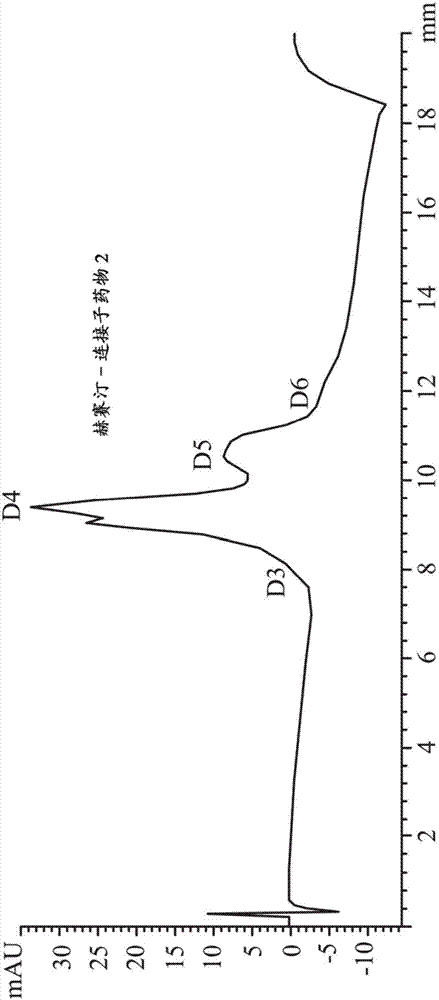
图2为根据本发明一实施例显示赫赛汀-连接子药物2的HIC图谱。
图3为根据本发明一实施例显示赫赛汀-连接子药物3的HIC图谱。
图4为根据本发明一实施例显示赫赛汀-连接子药物5的HIC图谱。
图5为根据本发明一实施例显示赫赛汀-连接子药物7的HIC图谱。
图6为根据本发明一实施例显示赫赛汀-连接子药物8的HIC图谱。
图7为根据本发明一实施例显示赫赛汀-连接子药物9的HIC图谱。
图8为根据本发明一实施例显示赫赛汀-连接子药物11的HIC图谱。
图9为根据本发明一实施例显示赫赛汀-连接子药物12的HIC图谱。
图10为根据本发明一实施例显示赫赛汀-连接子药物13的HIC图谱。
图11为根据本发明一实施例显示赫赛汀-连接子药物14的HIC图谱。
图12为根据本发明一实施例显示赫赛汀-连接子药物15的HIC图谱。
图13为根据本发明一实施例显示赫赛汀-连接子药物16的HIC图谱。
图14为根据本发明一实施例显示赫赛汀-连接子药物M1的HIC图谱。
图15为根据本发明一实施例显示赫赛汀-连接子药物7的储存稳定性测试结果。
图16为根据本发明一实施例显示经ADC(赫赛汀-连接子药物3、赫赛汀-连接子药物8、赫赛汀-连接子药物9)处理的MDA-MB-468细胞的细胞存活率。
图17为根据本发明一实施例显示经ADC(赫赛汀-连接子药物14、赫赛汀-连接子药物15、赫赛汀-连接子药物16)处理的MDA-MB-468细胞的细胞存活率。
图18A为根据本发明一实施例显示经ADC(赫赛汀-连接子药物2、赫赛汀-连接子药物4、赫赛汀-连接子药物8、赫赛汀-连接子药物9)处理的JIMT-1细胞的细胞存活率。
图18B为根据本发明一实施例显示经ADC(赫赛汀-连接子药物7)处理的JIMT-1细胞的细胞存活率。
图18C为根据本发明一实施例显示经ADC(赫赛汀-连接子药物11)处理的JIMT-1细胞的细胞存活率。
图19为根据本发明一实施例显示经ADC(赫赛汀-连接子药物14、赫赛汀-连接子药物15、赫赛汀-连接子药物16)处理的JIMT-1细胞的细胞存活率。
图20A为根据本发明一实施例显示经ADC(赫赛汀-连接子药物2、赫赛汀-连接子药物3、赫赛汀-连接子药物4)处理的BT-474细胞的细胞存活率。
图20B为根据本发明一实施例显示经ADC(赫赛汀-连接子药物8、赫赛汀-连接子药物9)处理的BT-474细胞的细胞存活率。
图20C为根据本发明一实施例显示经ADC(赫赛汀-连接子药物7、赫赛汀-连接子药物11)处理的BT-474细胞的细胞存活率。
图21为根据本发明一实施例显示经ADC(赫赛汀-连接子药物14、赫赛汀-连接子药物15、赫赛汀-连接子药物16)处理的BT-474细胞的细胞存活率。
图22为根据本发明一些实施例显示在BT-474异种移植模式中经ADC注射小鼠的肿瘤体积变化。
图23为根据本发明一实施例显示在EC PDX模式中经ADC注射小鼠的肿瘤体积变化。
实施方式
本发明提供了一种新颖的接合连接子(conjugating linker)结构,其具有两个连接点,其可以特异性地接合存在于抗原的两个半胱氨酸残基(链间二硫键完全/部分还原之后)的两个巯基(thiol group)。因此,由本发明提供的新型接合连接体结构所获得的抗体药物复合物(ADC)具有改善的结构稳定性。而且,与传统的抗体药物复合物相比,也获得了更窄的药物抗体比(drug to antibody ratio;DAR)分布。
所述新颖的接合连接子结构被设计为包含两个乙酰胺基团(acetamide groups)。两个乙酰胺基团用于与链间巯基特异性结合(链间双硫键还原后),形成类似于双硫键的结构,从而提供连接子和抗体之间的稳定结合。同时,所述接合连接子结构的另一部分,例如C-末端的官能基团可接合至药物单元或连接子药物单元以产生抗体药物复合物。
如此获得的抗体药物复合物可用于选择性地将药物递送至目标细胞,例如肿瘤细胞。抗体药物复合物将与细胞表面抗原特异性结合,并且此结合后的复合物将被细胞快速内化(intermalized)。一旦内化之后,药物将以一定的活性形式释放并发挥作用。与传统药物相比,本发明提供的抗体药物复合物不仅具有改良的结构稳定性,还具有改良的药学均质性。
定义
在以下描述中,术语“抗体”可以包括全长抗体或与受体、抗原、或其他目标细胞相关的部分结合、反应性相关、或复合的抗体片段。抗体可以是任何蛋白质、类蛋白质分子、或多肽,其与寻求治疗性修饰的细胞群的一部分结合、复合、或反应。
于一些实施例中,所述抗体可为嵌合抗体或其功能活性片段、人源化抗体或其功能活性片段、人类抗体或其功能活性片段。于其他实施例中,所述抗体亦可为上述物种以外的其他物种的抗体或其其功能活性片段,例如:小鼠抗体或其功能活性片段、大鼠抗体或其功能活性片段、羊抗体或其功能活性片段、兔子抗体或其功能活性片段。
于另一些实施例中,该抗体可为IgG1抗体或其功能活性片段、IgG4抗体或其功能活性片段。例如,抗体可为但不限于:HLX-07、EG12014、抗-EpCAM抗体、IgG1、利妥昔单抗(Rituximab)、替伊莫单抗(Ibritumomab tiuxetan)、托西莫单抗(Tositumomab)、本妥昔单抗(Brentuximab)、阿仑单抗(Alemtuzumab)、IGN101、阿德木单抗(Adecatumumab)、拉妥珠单抗(Labetuzumab)、huA33、Pemtumomab、Oregovomab、CC49(明瑞莫单抗;minretumomab)、cG250、J591、MOv18、Farletuzumab(MORAb-003)、3F8、ch14.18、KW-2871、hu3S193、IgN311、贝伐单抗(Bevacizumab)、IM-2C6、CDP791、伊瑞西珠单抗(Etaracizumab)、伏洛昔单抗(Volociximab)、西妥昔单抗(Cetuximab)、帕尼单抗(Panitumumab)、尼妥珠单抗(Nimotuzumab)、806、曲妥珠单抗(Trastuzumab)、帕妥珠单抗(Pertuzumab)、MM-121、AMG 102、METMAB、SCH 900105、AVE1642、IMC-A12、MK-0646、R1507、CP 751871、KB004、IIIA4、马帕木单抗(Mapatumumab)(HGS-ETR1)、来沙木单抗(Lexatumumab)(HGS-ETR2)、CS-1008、狄诺塞麦单抗(Denosumab)、西罗珠单抗(Sibrotuzumab)、F19、81C6、人源化抗HER2mAb、依决洛单抗(Edrecolomab)、西妥昔单抗(Cetuximab)、Smart MI95、LymphoCide、Smart ID10、Oncolym、Allomune、或依帕珠单抗(Epratuzamab)。
于一些实施例中,抗体可为多株抗体或单株抗体。于一些实施例中,抗体可为双特异性抗体。于一些实施例中,抗体也可为功能性活性片段、抗体的衍生物或类似物,其中该抗体可免疫特异性地结合至目标抗原(例如:癌症抗原、病毒抗原、微生物抗原、或其他可结合至细胞或基质的抗原)。在此,“功能性活性”代表所述片段、衍生物或类似物可辨认相同的抗原,辨认衍生自抗原的片段、衍生物或类似物的抗体。其他有用的抗体包括抗体的片段,例如但不限于:F(ab’)2片段、Fab片段、Fab’、Fv片段及抗体的重链及轻链二聚体、或其任何最小片段像是Fvs或单链抗体(SCAs)。
于其他实施例中,抗体可为抗体的融合蛋白、或其功能性活性片段。例如,抗体可通过非抗体的另一个蛋白质(或其部分,像是蛋白质的至少10、20或50个氨基酸)的氨基酸序列的N-端或C-端共价键进行融合(例如:肽键)。
于一些实施例中,抗体也可包括经修饰或未经修饰(亦即,通过任何分子的共价连接)的类似物和衍生物,只要这样的共价连接允许抗体保留其抗原结合免疫特异性。例如但不限于:抗体的类似物和衍生物,包括经过进一步修饰,例如:糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过习知的保护/阻隔基衍生化、蛋白酶切割、连接至细胞抗体单元或其他蛋白质等。可利用习知技术实现任何大量的化学修饰,包括但不限于特异性化学切割、乙酰化、甲酰化、衣霉素存在下的代谢合成等。此外,类似物或衍生物可包括一种或多种非天然氨基酸。于一些实施例中,抗体可在与Fc受体作用的氨基酸残基中具有修饰(例如:取代、删除、或增加)。
可用于治疗癌症的抗体例子包括但不限于:人源化抗HER2单株抗体、(trastuzumab;Genentech)、用于治疗具有非霍奇金氏淋巴瘤(non-Hodgkin's lymphoma)的病患的嵌合抗CD20单株抗体(rituximab;Genentech)、用于治疗卵巢癌的小鼠抗体OvaRex(AltaRex Corporation,MA)、用于治疗大肠直肠癌的小鼠IgG2a抗体Panorex(Glaxo Wellcome,NC)、用于治疗表皮生长因子阳性癌(像是头颈癌)的抗-EGFR IgG嵌合抗体Cetuximab Erbitux(Imclone Systems Inc.,NY)、用于治疗肉瘤(sarcoma)的人源化抗体Vitaxin(Medimmune,Inc.,MD)、用于治疗慢性淋巴球性白血病(chronic lymphocytic leukemia;CLL)的人源化IgG1抗体Campath I/H(Leukosite,MA)、用于治疗急性骨髓性白血病(acute myeloid leukemia;AML)的人源化抗-CD33IgG抗体Smart MI95(Protein Design Labs,Inc.,CA)、用于治疗非霍奇金氏淋巴瘤(non-Hodgkin's lymphoma)的人源化抗-CD22IgG抗体LymphoCide(Immunomedics,Inc.,NJ)、用于治疗非霍奇金氏淋巴瘤(non-Hodgkin's lymphoma)的人源化抗-HLA-DR抗体Smart ID10(Protein Design Labs,Inc.,CA)、用于治疗非霍奇金氏淋巴瘤(non-Hodgkin's lymphoma)的放射性标记小鼠抗-HLA-Dr10抗体Oncolym(Techniclone,Inc.,CA)、用于治疗霍奇金氏淋巴瘤(Hodgkin's lymphoma)或非霍奇金氏淋巴瘤(non-Hodgkin's lymphoma)的人源化抗-CD2单株抗体(mAb)Allomune(BioTransplant,CA)、用于治疗肺癌及大肠直肠癌的抗-VEGF人源化抗体Avastin(Genentech,Inc.,CA)、用于治疗非霍奇金氏淋巴瘤(non-Hodgkin's lymphoma)的抗-CD22抗体Epratuzamab(Immunomedics,Inc.,NJ and Amgen,CA)、及用于治疗大肠直肠癌的人源化抗-CEA抗体CEAcide(Immunomedics,NJ)。
其他可用于治疗癌症的抗体可包括但不限于对抗下述抗原的抗体(在括号中指出示例性癌症):CA125(卵巢癌)、CA15-3(癌;carcinomas)、CA19-9(癌)、L6(癌)、Lewis Y(癌)、Lewis X(癌)、α-胎儿蛋白(癌)、CA 242(大肠直肠癌)、胎盘碱性磷酸酶(癌)、前列腺癌特异性膜抗原(前列腺癌)、前列腺酸性磷酸酶(前列腺癌)、表皮生长因子(癌)、MAGE-1(癌)、MAGE-2(癌)、MAGE-3(癌)、MAGE-4(癌)、抗输铁蛋白受体(癌)、p97(黑色素瘤)、MUC1-KLH(乳癌)、CEA(大肠直肠癌)、gp100(黑色素瘤)、MART1(黑色素瘤)、前列腺特异性抗体(PSA)(前列腺癌)、IL-2受体(T-细胞白血病和淋巴瘤)、CD20(非霍奇金氏淋巴瘤)、CD52(白血病)、CD33(白血病)、CD22(淋巴瘤)、人绒毛膜促性腺激素(癌)、CD38(多发性骨髓瘤)、CD40(淋巴瘤)、黏蛋白(癌)、P21(癌)、MPG(黑色素瘤)、及Neu癌基因产物(癌)。
于一些实施例中,抗体对于自体免疫疾病的治疗具有免疫特异性,例如:抗-双股DNA、抗-单股DNA、抗-心磷脂抗体IgM或IgG、抗-磷脂抗体IgM或IgG、抗-SM抗体、抗-粒线体抗体、甲状腺抗体、微粒体抗体、甲状腺球蛋白抗体、抗-SCL 70、抗-Jo、抗-U1RNP、抗-La/SSB、抗-SSA、抗-SSB、抗-壁细胞抗体(anti-periteal cells)、抗-组蛋白、抗-RNP、C ANCA、P ANCA、抗中心粒、抗纤维蛋白、及抗-GBM抗体。于一实施例中,结合至活化淋巴球的配体(ligand)与自体免疫疾病相关。
于特定实施例中,抗体可结合至表现在目标细胞上的抗体或抗体复合物(例如:活化的淋巴球)。抗体或抗体复合物可包括免疫球蛋白基因超家族成员、TNF受体超家族成员、整合蛋白(integrin)、细胞激素受体、化学激素受体、主要组织相容性复合物、红血球凝集素(lectin)、或补体控制蛋白。合适的免疫球蛋白基因超家族成员包括但不限于:CD2、CD3、CD4、CD8、CD19、CD22、CD28、CD79、CD90、CD152/CTLA 4、PD 1、及ICOS。合适的TNF受体超家族成员包括但不限于:CD27、CD40、CD95/Fas、CD134/OX40、CD137/4 1BB、TNF R1、TNFR2、RANK、TACI、BCMA、蚀骨细胞抑制因子(osteoprotegerin)、Apo2/TRAIL R1、TRAIL R2、TRAIL R3、TRAIL R4、及APO 3。合适的整合蛋白(integrin)包括但不限于:CD11a、CD11b、CD11c、CD18、CD29、CD41、CD49a、CD49b、CD49c、CD49d、CD49e、CD49f、CD103、及CD104。合适的红血球凝集素(lectin)包括但不限于:C型、S型、及I型红血球凝集素。
抗体也可为存在于目标细胞或目标细胞群上的抗体。例如:相较于一个或多个正常细胞(例如:非癌细胞),可特异性表现在一个或多个特定类型的目标细胞(例如:癌细胞)表面上的跨膜多肽及其他标记分子。相较于正常细胞表面上的标记分子,这些标记分子经常丰富表现在目标细胞表面上、或显现更佳的免疫原性。这种细胞表面抗原多肽的辨识通过抗体疗法对特定的目标细胞具有破坏能力。因此,于一些实施例中,抗体包括但不限于对抗肿瘤相关抗原(TAA)的抗体。
在以下描述中,术语“药物单元”或“D”是指具有期望的生物学活性和反应性官能团的任何化合物,其可用于将药物并入本发明的复合物。于一些实施例中,药物单元表示用于癌症治疗的细胞毒性药物;具有所需生物活性的蛋白质或多肽,例如毒素像是相思豆毒蛋白、蓖麻毒蛋白A、假单胞菌外毒素、和白喉毒素;其它合适的蛋白质,包括肿瘤坏死因子、α-干扰素、β-干扰素、神经生长因子、血小板衍生的生长因子、组织纤溶酶原激活剂、和生物反应调节剂,例如:淋巴因子、白细胞介素-1(IL-1)、白细胞介素-2(IL-2)、白细胞介素-6(IL-6)、粒细胞巨噬细胞集落刺激因子(GM-CSF)、粒细胞集落刺激因子(G-CSF)、或其他生长因子。
于一实施例中,药物单位可以是微管干扰药物,例如澳瑞他汀,像是单甲基澳瑞他汀E(monomethyl auristatin E;MMAE)、单甲基澳瑞他汀F(monomethyl auristatin F;MMAF)、和澳瑞他汀F(auristatin F;AF)。于另一实施例中,药物单元可以是微管破坏药物,例如美登木素生物碱,像是DM1、DM3、和DM4。于另一实施例中,药物单元可以是DNA损伤剂,像是加利车霉素(calicheamicins)、多卡霉素(duocarmycins)、SN-38、和吡咯并[2,1-c][1,4]苯并二氮杂(pyrrolo[2,1-c][1,4]benzodi-azepines;PBDs)。又于另一实施例中,药物单元可以是瓢菌素(amanitins)、蒽环类物(anthracyclines)、浆果赤霉素(baccatins)、喜树碱(camptothecins)、西马多丁(cemadotins)、秋水仙碱(colchicines)、秋水仙胺(colcimids)、考布他汀(combretastatins)、隐菲辛(cryptophycins)、圆皮海绵内酯(discodermolides)、多烯紫杉醇(docetaxel)、阿霉素(doxorubicin)、棘霉素(echinomycins)、艾榴塞洛素(eleutherobins)、埃博霉素(epothilones)、雌莫司汀(estramustines)、偏端霉素(lexitropsins)、美登素(maytansines)、氨甲蝶呤(methotrexate)、纺锤菌素(netropsins)、嘌呤霉素(puromycins)、根瘤菌素(rhizoxins)、紫杉烷(taxanes)、微管蛋白裂解素(tubulysins)、或长春花生物碱(vinca alkaloids)。
应注意的是,药物不限于上述类别,并包括所有可用于ADC的药物。
在以下描述中,本发明中使用的术语“连接单元”包括可切割的连接子或不可切割的连接子。可切割的连接子可为化学不稳定的和酶不稳定的连接子。由于高血浆稳定性和良好的细胞内切割选择性和效率,在ADC中广泛选择酶不稳定连接子作为可切割连接子候选物。于一些实施例中,酶不稳定连接子可包括肽单元(-AAs-),选自于由-缬氨酸-瓜氨酸-(-Val-Cit-)、-缬氨酸-赖氨酸-(-Val-Lys-)、-缬氨酸-精氨酸-(-Val-Arg-)、-苯丙氨酸-瓜氨酸-(-Phe-Cit-)、-苯丙氨酸-赖氨酸-(-Phe-Lys-)、和-苯丙氨酸-精氨酸-(-Phe-Arg-)所组成的群组。典型的酶不稳定连接子包括-Val-Cit-和-Phe-Lys-,其可被组织蛋白酶B识别。于一些实施例中,不可切割的连接子可为能够增加所得ADC的亲水性的连接子。于一实施例中,不可切割的连接子可包括一或多个聚(乙二醇)(PEG)。于另一实施例中,不可切割的连接子可为PEG、PEG二胺(NH2-PEG-NH2)、胺-PEG-羟基(NH2-PEG-OH)、胺-PEG-COOH(NH2-PEG-COOH)、二亚乙基三胺、或前述的组合。于一些实施例中,PEG可如所示,其中x可为从1至20的整数。
在以下描述中,本发明中使用的术语“接合连接子”指用于与抗体接合的连接子的新颖接合连接子结构。以下段落将详细描述接合连接子。
于一实施例中,本发明提供一种如化学式(I)所示的连接子药物:
C-(L-D)m (I)
于化学式(I)中,C为接合连接子,L为连接子单元,D为药物单元,且m为从1至4的整数。接合连接子(-C)的结构如化学式(II)所示:
于化学式(II)中,X为离去基。于一些实施例中,离去基(-X)可为-Cl、-Br、-I、-F、-OTs、-OMs、-OTf、或-OBs。于化学式(II)中,R1和R2同时为单键或–NH–,Z为芳基、经取代的芳基、杂芳基、经取代的杂芳基、直链烷基、环烷基、杂环烷基、或前述的组合。化学式(II)中的波浪线表示与L的共价连接位。
于化学式(II)中,R1和R2彼此以邻位、间位、或对位的方式连接至Z。其中,Z系如化学式(III)所示:
于化学式(III)中,Q为取代基,包括H、硝基、氰基、羟基、烷氧基、胺基、酰胺基、酯基、磺酰胺基、脲基、直链烷基、环烷基、杂环烷基、烯基、炔基、芳基、杂芳基;n为从0至3的整数;其中,一或多个苯环上的–CH–未被取代或是被N取代。
于一些实施例中,接合连接子(-C)的结构系如化学式(IIa)、化学式(IIb)、化学式(IIc)、或化学式(IId)所示:
然而,应该理解的是,上述结构仅仅是示例,本发明的范围并不限于此。
于一些实施例中,连接子单元(-L)可包括可切割的连接子或不可切割的连接子。于一些实施例中,可切割的连接子可包括肽单元(-AAs-),选自于由-缬氨酸-瓜氨酸-(-Val-Cit-)、-缬氨酸-赖氨酸-(-Val-Lys-)、-缬氨酸-精氨酸-(-Val-Arg-)、-苯丙氨酸-瓜氨酸-(-Phe-Cit-)、-苯丙氨酸-赖氨酸-(-Phe-Lys-)、和-苯丙氨酸-精氨酸-(-Phe-Arg-)所组成的群组。于一些实施例中,肽单元(-AAs-)可被一种或多种酶(例如肿瘤相关蛋白酶)切割以释放药物单元(-D)。于一些实施例中,不可切割的连接子可为能够增加所得ADC的亲水性的连接子。于一实施例中,不可切割的连接子可包括一或多个聚(乙二醇)(PEG)。于另一实施例中,不可切割的连接子可为PEG、PEG二胺(NH2-PEG-NH2)、胺-PEG-羟基(NH2-PEG-OH)、胺-PEG-COOH(NH2-PEG-COOH)、二亚乙基三胺、或前述的组合。于一些实施例中,PEG可如所示,其中x可为从1至20的整数。
于一些实施例中,药物单元(-D)还可包括作为连接单元(-L)的连接位点的-脯氨酸-(-Pro-)。
于化学式(I)中,-(L-D)系如化学式(IIIa)或化学式(IIIb)所示:
L为直链烷基、环烷基、杂环烷基、烯基、炔基、芳基、杂芳基、聚(乙二醇)链、或前述的组合。每一个D独立地为细胞毒性药物、抗自体免疫疾病药物、或抗发炎药物;o为从1至4的整数;p为从1至4的整数;且q为从1至4的整数。当-(L-D)系如化学式(IIIb)所示时,其药物接合数量为如化学式(IIIb)所示-(L-D)的药物接合数量的两倍。应理解的是,根据设计,可能存在-(L-D)的其它变化以提供具有更多接合数量的药物的-(L-D)。
于一些实施例中,药物单元(-Do)还包括-脯氨酸-(-Pro-)作为连接单元(-L)的连接位点。于一些实施例中,药物单元(-Dp)还包括-脯氨酸-(-Pro-)作为连接单元(-L)的连接位点。于一些实施例中,药物单元(-Dq)还包括-脯氨酸-(-Pro-)作为连接单元(-L)的连接位点。
于化学式(I)中,药物单元可包括:瓢菌素(amanitins)、蒽环类物(anthracyclines)、澳瑞他汀(auristatins)、浆果赤霉素(baccatins)、加利车霉素(calicheamicins)、喜树碱(camptothecins)、西马多丁(cemadotins)、秋水仙碱(colchicines)、秋水仙胺(colcimids)、考布他汀(combretastatins)、隐菲辛(cryptophycins)、圆皮海绵内酯(discodermolides)、多卡霉素(duocarmycins)、多烯紫杉醇(docetaxel)、阿霉素(doxorubicin)、多卡霉素(duocarmycins)、棘霉素(echinomycins)、艾榴塞洛素(eleutherobins)、埃博霉素(epothilones)、雌莫司汀(estramustines)、偏端霉素(lexitropsins)、美登素(maytansines)、美登木素生物碱(maytansinoids)、氨甲蝶呤(methotrexate)、纺锤菌素(netropsins)、吡咯并[2,1-c][1,4]苯并二氮杂(pyrrolo[2,1-c][1,4]benzodi-azepines;PBDs)、嘌呤霉素(puromycins)、根瘤菌素(rhizoxins)、SN-38、紫杉烷(taxanes)、微管蛋白裂解素(tubulysins)、或长春花生物碱(vinca alkaloids)。
于一实施例中,本发明提供一种如化学式(IV)所示的抗体药物复合物(ADC):
A-C’-(L-D)m (IV)
于化学式(IV)中,A为全长抗体、抗体片段、蛋白质、或多肽;C’-(L-D)m为连接子药物,其中C’为接合连接子;L为连接子单元;D为药物单元;且m为从1至4的整数。A通过分别存在于A的两个半胱氨酸残基中的两个巯基(thiol groups)与连接子药物接合。
于化学式(IV)中,A-C’包括如化学式(V)所示的下列结构:
于化学式(V)中,R1和R2同时为单键或–NH–;Z为芳基、经取代的芳基、杂芳基、经取代的杂芳基、直链烷基、环烷基、杂环烷基、或前述的组合。化学式(V)的波浪线表示与L的共价连接位。A’表示A的剩余部分,其通过分别存在于A的两个半胱氨酸残基中的两个巯基(thiol groups)与该连接子药物接合。
于化学式(IV)中,“A”靶向细胞表面受体或肿瘤相关抗原。于一些实施例中,抗体可为嵌合抗体或其功能活性片段、人源化抗体或其功能活性片段、人类抗体或其功能活性片段、小鼠抗体或其功能活性片段、大鼠抗体或其功能活性片段、羊抗体或其功能活性片段、或兔子抗体或其功能活性片段。
于一些实施例中,抗体为IgG1抗体或其功能活性片段、IgG4抗体或其功能活性片段。
于一些实施例中,抗体可包括:HLX-07、EG12014、抗-EpCAM抗体、IgG1、利妥昔单抗(Rituximab)、替伊莫单抗(Ibritumomab tiuxetan)、托西莫单抗(Tositumomab)、本妥昔单抗(Brentuximab)、阿仑单抗(Alemtuzumab)、IGN101、阿德木单抗(Adecatumumab)、拉妥珠单抗(Labetuzumab)、huA33、Pemtumomab、Oregovomab、CC49(明瑞莫单抗;minretumomab)、cG250、J591、MOv18、Farletuzumab(MORAb-003)、3F8、ch14.18、KW-2871、hu3S193、IgN311、贝伐单抗(Bevacizumab)、IM-2C6、CDP791、伊瑞西珠单抗(Etaracizumab)、伏洛昔单抗(Volociximab)、西妥昔单抗(Cetuximab)、帕尼单抗(Panitumumab)、尼妥珠单抗(Nimotuzumab)、806、曲妥珠单抗(Trastuzumab)、帕妥珠单抗(Pertuzumab)、MM-121、AMG 102、METMAB、SCH 900105、AVE1642、IMC-A12、MK-0646、R1507、CP 751871、KB004、IIIA4、马帕木单抗(Mapatumumab)(HGS-ETR1)、来沙木单抗(Lexatumumab)(HGS-ETR2)、CS-1008、狄诺塞麦单抗(Denosumab)、西罗珠单抗(Sibrotuzumab)、F19、81C6、人源化抗HER2mAb、依决洛单抗(Edrecolomab)、西妥昔单抗(Cetuximab)、Smart MI95、LymphoCide、Smart ID10、Oncolym、Allomune、或依帕珠单抗(Epratuzamab)。
于化学式(V)中,R1和R2彼此以邻位、间位、或对位的方式连接至Z,其中Z系如化学式(III)所示:
Q为取代基,包括H、硝基、氰基、羟基、烷氧基、胺基、酰胺基、酯基、磺酰胺基、脲基、直链烷基、环烷基、杂环烷基、烯基、炔基、芳基、杂芳基;n为从0至3的整数;其中,一或多个苯环上的–CH–未被取代或是被N取代。
于化学式(V)中,A-C’包括如化学式(Va)、化学式(Vb)、化学式(Vc)、或化学式(Vd)所示的下列结构:
然而,应该理解的是,上述结构仅仅是示例,本发明的范围并不限于此。
于一些实施例中,连接子单元(-L)可包括可切割的连接子或不可切割的连接子。于一些实施例中,可切割的连接子可包括肽单元(-AAs-),选自于由-缬氨酸-瓜氨酸-(-Val-Cit-)、-缬氨酸-赖氨酸-(-Val-Lys-)、-缬氨酸-精氨酸-(-Val-Arg-)、-苯丙氨酸-瓜氨酸-(-Phe-Cit-)、-苯丙氨酸-赖氨酸-(-Phe-Lys-)、和-苯丙氨酸-精氨酸-(-Phe-Arg-)所组成的群组。于一些实施例中,肽单元(-AAs-)可被一种或多种酶(例如肿瘤相关蛋白酶)切割以释放药物单元(-D)。于一些实施例中,不可切割的连接子可为能够增加所得ADC的亲水性的连接子。于一实施例中,不可切割的连接子可包括一或多个聚(乙二醇)(PEG)。于另一实施例中,不可切割的连接子可为PEG、PEG二胺(NH2-PEG-NH2)、胺-PEG-羟基(NH2-PEG-OH)、胺-PEG-COOH(NH2-PEG-COOH)、二亚乙基三胺、或前述的组合。于一些实施例中,PEG可如所示,其中x可为从1至20的整数。
于一些实施例中,药物单元(-D)还可包括作为连接单元(-L)的连接位点的-脯氨酸-(-Pro-)。
于化学式(IV)中,-(L-D)系如化学式(IIIa)或化学式(IIIb)所示:
L为直链烷基、环烷基、杂环烷基、烯基、炔基、芳基、杂芳基、聚(乙二醇)链、或前述的组合。每一个D独立地为细胞毒性药物、抗自体免疫疾病药物、或抗发炎药物;o为从1至4的整数;p为从1至4的整数;且q为从1至4的整数。当-(L-D)系如化学式(IIIb)所示时,其药物接合数量为如化学式(IIIb)所示-(L-D)的药物接合数量的两倍。应理解的是,根据设计,可能存在-(L-D)的其它变化以提供具有更多接合数量的药物的-(L-D)。
于一些实施例中,药物单元(-Do)还包括-脯氨酸-(-Pro-)作为连接单元(-L)的连接位点。于一些实施例中,药物单元(-Dp)还包括-脯氨酸-(-Pro-)作为连接单元(-L)的连接位点。于一些实施例中,药物单元(-Dq)还包括-脯氨酸-(-Pro-)作为连接单元(-L)的连接位点。
于化学式(I)中,药物单元可包括:瓢菌素(amanitins)、蒽环类物(anthracyclines)、澳瑞他汀(auristatins)、浆果赤霉素(baccatins)、加利车霉素(calicheamicins)、喜树碱(camptothecins)、西马多丁(cemadotins)、秋水仙碱(colchicines)、秋水仙胺(colcimids)、考布他汀(combretastatins)、隐菲辛(cryptophycins)、圆皮海绵内酯(discodermolides)、多卡霉素(duocarmycins)、多烯紫杉醇(docetaxel)、阿霉素(doxorubicin)、多卡霉素(duocarmycins)、棘霉素(echinomycins)、艾榴塞洛素(eleutherobins)、埃博霉素(epothilones)、雌莫司汀(estramustines)、偏端霉素(lexitropsins)、美登素(maytansines)、美登木素生物碱(maytansinoids)、氨甲蝶呤(methotrexate)、纺锤菌素(netropsins)、吡咯并[2,1-c][1,4]苯并二氮杂(pyrrolo[2,1-c][1,4]benzodi-azepines;PBDs)、嘌呤霉素(puromycins)、根瘤菌素(rhizoxins)、SN-38、紫杉烷(taxanes)、微管蛋白裂解素(tubulysins)、或长春花生物碱(vinca alkaloids)。
理论上,DAR值对应于还原之后还原抗体所提供的游离硫醇的数量。例如,在抗体中的四个链间二硫键被转化为8个游离巯基后,位点特异性接合连接子可以与存在于抗原半胱氨酸残基中的8个游离巯基特异性接合。因此,由本发明提供的新颖接合连接子结构所获得的抗体药物复合物(ADC)的DAR值约为4,而连接子单元(-L)接合至1个药物单位(-D)。于另一实施例中,由本发明提供的新颖接合连接子结构所获得的抗体药物复合物(ADC)的DAR值约为8,而连接子单元(-L)接合至2个药物单位(-D)。
以下提供实施例和比较例说明连接子药物和抗体药物复合物的形成方法,以及抗体药物复合物的特性:
实施例与比较例
以下,将通过实施例来详细描述本发明。其中,MMAE和AF系购自于Concortis Biotherapeutics。上述化合物的结构为本领域具有通常知识者所熟知,为达简明的目的,此处不再赘述。
表1中列出了连接子药物的缩写及其相应的化学结构。
表1
实施例1:连接子药物1的制备
连接子药物1系根据以下方案所示的步骤合成。
步骤1:合成化合物1a(Compound 1a)
化合物1a的合成如专利文献US9086416B2所述实施。3,5-二氨基苯甲酸(1.52g,10mmole)和二叔丁氧基二碳酸酯((t-Boc)2O;6.55g,30mmole)与三乙胺(NEt3;7.0mL,60mmole)在CH2Cl2(200mL)中,于室温(25℃)搅拌过夜(24小时)。将得到的溶液用水萃取,用Na2SO4干燥以得到褐色粉末的化合物1a(N-Boc-3,4-二氨基苯甲酸)(1.37g,产率54%)。
化合物1a的化学式:C17H24N2O6。1H NMR(500MHz,CDCl3)δ8.04(s,1H,ArH),7.91-7.89(dd,2H,ArH),7.23(s,1H),6.60(brs,2H,NH),1.54(s,18H,CH3)。M/Z(ES+):354.0(M+H)+,376.0(M+Na)。
步骤2:合成化合物1b(Compound 1b)
于化合物1a(60mg,0.17mmole)和五氟苯酚(38mg;0.20mmole)在DCM(6mL)中的混合物溶液添加DCC(42mg,0.20mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。以正己烷/EtOAc(4:1)洗脱管柱。收集含目标化合物的部分,减压浓缩以得到化合物1b(76mg,0.14mmole,产率80%)。
化合物1b的化学式:C23H23F5N2O6。1H-NMR(500MHz;CDCl3)d:8.12(s;1H);7.97(s;2H),7.25(s;1H),1.5(s,18H),MS(ESI,阴离子):M/Z:517.9(M-)。
步骤3:合成化合物1c(Compound 1c)
于化合物1b(11.6mg,0.22mmole)在DCM(1mL)和DMF(2mL)中的溶液添加L1(55mg,0.33mmole)、DIPEA(47.4mg,0.55mmole)。反应在室温下放置1小时。除去溶剂后,通过制备型HPLC纯化化合物1c(50%CH3CN的H2O溶液+0.1%TFA;UV 210nm;Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物1c,停留时间14.6min)。获得白色固体化合物1c(48mg;产率57%)。
化合物1c的化学式:C27H44N3O11。ESI-HIMS(阳离子),M/Z:586.2972,[M+H]+for C27H44N3O11,err:-0.3ppm。
步骤4:合成化合物1d(Compound 1d)
于化合物1c(28mg,0.04mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和MMAE(60mg,0.07mmole)在2mL DMF中的溶液添加TBTU(44mg)和DIPEA(36mg)。4小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物1d,停留时间17min)。获得白色固体化合物1d(42mg;产率70%)。
化合物1d的化学式:C66H108N8O17。(ESI,阳离子):M/Z:1286.9,(M+1);1308.8(M+Na)。
步骤5:合成化合物1(Compound 1)
在25~27℃于化合物1d(62mg,0.048mmole)在2mL DCM中的溶液滴加1mL(11.5mmole)的三氟乙酸。接着除去水浴并继续搅拌4小时。减压除去溶剂,于残余物中加入5mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(1.5mL)并冷冻干燥以得到粗产物化合物1d-deBoc 49mg做为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐(49mg,0.048mmole)的DCM(1.5mL)溶液添加三甲胺(45μL,0.613mmole),随后加入氯乙酰氯化物43mg(0.377mmole)的DCM(0.3mL)溶液。搅拌后,加入第二部分三甲胺(45μL,0.613mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.43%梯度至含有0.1%TFA的55%AcN/H2O,UV220nm)。将含产物的部分(Rt=16.1min)冷冻干燥以得到化合物1,即连接子药物1(43mg,产率68.3%)。
连接子药物1的化学式:C80H94Cl2N8O5。(ESI,阳离子):M/Z:[M+H]+=1238.7,[M+Na]+=1260.7。
实施例2:连接子药物2的制备
连接子药物2系根据以下方案所示的步骤合成。
步骤1~4:合成化合物2a~2d(Compounds 2a~2d)
通过与实施例1中所述步骤1~4相同的合成步骤合成化合物2a、2b、2c、和2d。
步骤5:合成化合物2(Compound 2)
在20~23℃于化合物2d(40mg,0.031mmole)在1mL DCM中的溶液滴加0.8mL(9.12mmole)的三氟乙酸。除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物2d-deBoc 27mg做为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐(27mg,0.031mmole)的DCM(1mL)溶液添加三甲胺(20μL,0.136mmole)的DCM(0.3mL)溶液,随后加入溴乙酰溴化物42mg(0.208mmole)的DCM(0.3mL)溶液。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以4mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.43%梯度至含有0.1%TFA的62%AcN/H2O,UV220nm)。将含产物的部分(Rt=17~19min)冷冻干燥以得到化合物2,即连接子药物2(2.6mg,产率6.8%)。
连接子药物2的化学式:C60H84Br2N8O15。(ESI,阳离子):M/Z:[M+H]+=1326.8。
实施例3:连接子药物3的制备
连接子药物3系根据以下方案所示的步骤合成。
步骤1:合成化合物3a(Compound 3a)(N-Boc-3,6-二氧辛烷-1,8-二胺)
化合物DAB-2按照文献方法(Bioorganic)制备,且将二叔丁基二碳酸酯((Boc)2O)(0.7g,3.21mmol)的CH2Cl2(10mL)溶液滴加至三(乙二醇)-1,8-二胺(5.0g,278mmol)和二异丙基乙胺(10mL,57mmol)的混合物中,于室温经过2小时。将反应混合物搅拌6小时,然后将其真空浓缩。通过快速硅胶管柱层析(CH2Cl2/CH3OH,10/1,v/v)纯化,得到N-Boc-3,6-二氧杂辛烷-1,8-二胺(0.6g,75.6%)。1H NMR(500MHz,CDCl3):δ3.61(s,4H),3.55-3.53(t,2H),3.52-3.49(t,2H),3.31-3.30(t,2H),2.88-2.85(t,2H),1.43(s,9H)。
步骤2:合成化合物3b(Compound 3b)(Boc-PEG-AF)
于澳瑞他汀F(200mg,0.23mmole)在DCM(2mL)和DMF(2mL)中的溶液添加化合物3a(75mg,0.30mmole)、DIPEA(180mg,1.39mmole)、TBTU(220mg,0.68mmole)。接着将反应在室温下搅拌1小时。除去溶剂后,通过制备性HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流动相43%AcN/H2O,含有0.1%TFA,流速32mL/min;Boc-PEG-AF,室温9.3分钟)。获得白色固体化合物3b(203mg;89%)。ESI,阳离子:M/Z:977.5(M+1);993.2(M+Na)。
步骤3:合成化合物3c(Compound 3c)
向化合物3b(203mg,0.21mmol)的DCM(4mL)溶液中添加TFA(2mL)。将反应混合物在室温搅拌1小时,此后在真空下除去溶剂。粗产物不经进一步纯化而使用。ESI,阳离子:M/Z:877.4(M+1);899.4(M+Na)。
步骤4:合成化合物3(Compound 3)
于化合物3c(180mg,0.20mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和化合物3,4-双(2-氯乙酰胺基)苯甲酸(62mg,0.20mmole)在2mL DMF中的溶液添加TBTU(191mg)和DIPEA(154mg)。反应于4小时后结束,接着减压除去溶剂。通过RP-HPLC(Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物3,停留时间17min)进行纯化。获得白色固体化合物3,即连接子药物3(167mg;70%)。
连接子药物3的化学式:C57H89Cl2N9O12。ESI,阳离子:M/Z:1163.5(M+1);1185.3(M+Na).ESI-HIMS,阳离子:M/Z:1162.6082,[M+H]+for C57H90Cl2N9O12,err:-0.1ppm。
实施例4:连接子药物4的制备
连接子药物4系根据以下方案所示的步骤合成。
步骤1~3:合成化合物4a~4c(Compounds 4a~4c)
通过与实施例3中所述步骤1~3相同的合成步骤合成化合物4a、4b、和4c。
步骤4:合成化合物4(Compound 4)
在20~23℃水浴中,于化合物4b(90mg,0.074mmole)在4mL DCM中的溶液滴加2mL(10.4mmole)的三氟乙酸。接着除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物4c 90mg作为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐(90mg,0.074mmole)的DCM(1.5mL)溶液添加三甲胺(26μL,0.18mmole),随后加入溴乙酰溴化物64mg(0.31mmole)的DCM(0.3mL)溶液。搅拌5分钟后,加入第二部分三甲胺(52μL,0.36mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.43%AcN/H2O含有0.1%TFA,UV220nm)。将含产物的部分(Rt=7.66min)冷冻干燥以得到化合物4,即连接子药物4(85mg,产率92%)。
连接子药物4的化学式:C57H89Br2N9O12。(ESI,阳离子):M/Z:M+H=1251.6。
实施例5:连接子药物5的制备
连接子药物5系根据以下方案所示的步骤合成。
步骤1:合成化合物5a(Compound 5a)
化合物5a也称为(亚氨基二乙烷-2,1-二基)双胺甲酸二叔丁酯(DETA-3)。根据文献方法(Org.Lett.2000,2,2177-2120)制备化合物5a。
首先,按照以下的方法合成DETA-2。将二乙烯三胺(0.82g,6.2mmol)和三乙胺(2.6mL,8.6mmol,3当量)溶于四氢呋喃(40mL)中并在冰浴中冷却。然后,经2.5小时滴加Boc2O(3.1g,12.4mmol,2当量)的四氢呋喃(20mL)溶液。接着将反应混合物在0℃再搅拌1小时,然后让其达到环境温度过夜。接着,将反应混合物真空浓缩。将残余物溶于二氯甲烷中,并用1M的氢氧化钠水溶液洗涤。用Na2SO4干燥有机相并真空浓缩。通过快速色谱纯化粗产物,得到(亚氨基二乙烷-2,1-二基)双胺甲酸二叔丁酯(DETA-2,M/Z(ES-),302.9(M-H)-)和DETA-2-1(M/Z-(ES-),403.0(M-H)-)的黄色粘稠油状物。向化合物DETA-2和DETA-2-1(0.86g)的DCM(20mL)溶液中添加琥珀酸酐(0.3g,3mmol)。将反应混合物在室温搅拌16小时。在真空下除去溶剂,并通过管柱层析法纯化残余物(硅胶,DCM/MeOH=20/1)以得到白色固体(0.43g)。1H NMR(500MHz,CDCl3)δ5.21(br,1H),4.98(br,1H),3.32(br,6H),3.24(bs,6H),1.45(s,9H,CH3),1.46(s,9H,CH3),1.42(s,9H,CH3);M/Z(ES+),404.9(M+H),426.7(M+Na)。
接下来,于化合物DETA-2(0.86g,2.84mmole)的DCM(20mL)溶液中添加琥珀酸酐(0.3g,3mmol)。接着将反应混合物在室温下搅拌16小时。在真空下除去溶剂,并通过管柱层析法纯化残余物(硅胶,DCM/MeOH=20/1)以得到白色固体化合物5a(0.43g)。1H NMR(500MHz,CDCl3)δ5.21(br,1H),4.98(br,1H),3.68-3.71(m,4H),3.46-3.48(m,4H),2.62-2.69(m,4H),1.45(s,18H,CH3)MS:(ESI+)M/Z,404.9(M+H)+.426.2(M+Na)。
步骤2:合成化合物5b(Compound 5b)
于化合物5a(170mg,0.42mmole)和五氟苯酚(93mg;0.50mmole)在DCM(6mL)中的混合物溶液添加DCC(104mg,0.50mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。以正己烷/EtOAc(4:1)洗脱管柱。收集含目标化合物的部分,减压浓缩以得到化合物5b(168mg,0.29mmole,产率70%)。MS(ESI,阴离子):M/Z:570.9(M+1)+,592.8(M+Na)+。
步骤3:合成化合物5c(Compound 5c)
于化合物5b(168mg,0.29mmole)在DCM(2mL)和DMF(2mL)中的溶液添加CJ35-5(111mg,0.43mmole)、DIPEA(95mg,0.50mmole)。反应在室温下放置2小时。除去溶剂后,通过制备型HPLC纯化化合物5c(50%CH3CN的H2O溶液+0.1%TFA;UV 210nm;Inertsil ODS-3管柱30x250mm;流速31mL/min;化合物5c,停留时间6.9min)。获得白色固体化合物5c(125mg;产率67%)。MS(ESI,阴离子):M/Z:635.7(M-H)+。
步骤4:合成化合物5d(Compound 5d)
于化合物5c(125mg,0.04mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和化合物MMAE(141mg,0.07mmole)在2mL DMF中的溶液添加TBTU(183mg)和DIPEA(147mg)。4小时后反应完成。接着减压除去溶剂。Inertsil ODS-3管柱30x250mm;流动相AcN/H2O4 1%梯度至50%,流速32mL/min;化合物5d,停留时间23.7min)。获得白色固体化合物5d(186mg;产率71%)。ESI-MS(ESI,阳离子):M/Z:1338.0(M+H)+,1359.9(M+Na)+。
步骤5:合成化合物5(Compound 5)
在5~8℃于化合物5d(92mg,0.068mmole)在1mL DCM中的溶液滴加0.8mL(10.4mmole)的三氟乙酸。接着除去水浴并继续搅拌4小时。随后减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(3mL)并冷冻干燥以得到粗产物化合物5d-deBoc 62mg做为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐(62mg,0.054mmole)的DCM(1.7mL)溶液添加三甲胺(40μL,0.272mmole),随后加入溴乙酰溴化物60mg(0.297mmole)的DCM(0.3mL)溶液。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以4mL移动相溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,含0.1%TFA的流动相AcN/H2O梯度从40%至60%,化合物5,即连接子药物5,停留时间11.08min,UV 215nm)。将含产物的部分冷冻干燥以得到化合物5,即连接子药物5(15mg)。
连接子药物5的化学式:C61H103Br2N9O16。(ESI,阳离子):M/Z:[M+H]+=1377.6。
实施例6:连接子药物6的制备
连接子药物6系根据以下方案所示的步骤合成。
步骤1~4:合成化合物6a~6d(Compounds 6a~6d)
通过与实施例5中所述步骤1~4相同的合成步骤合成化合物6a、6b、6c、和6d。
步骤5:合成化合物6(Compound 6)
将化合物5(7.9mg,5.7μmol)和碘化钠(8.6g,57.3μmol)在乙腈(2mL)中的混合物在室温下搅拌8小时,将溶剂真空蒸发。以RP-HPLC进行纯化(43%CH3CN的H2O溶液,0.1%TFA,UV 220nm;Inertsil ODS-3管柱20x250mm;流速19mL/min,化合物6,即连接子药物6,停留时间23.2min)。获得白色固体连接子药物6(6.7mg;4.5μmol,产率80%)。
连接子药物6的化学式:C61H103I2N9O1。ESI-HIMS(阳离子)M/Z:1473.6,(M+H)+1495.5(M+Na)+。
实施例7:连接子药物7的制备
连接子药物7系根据以下方案所示的步骤合成。
步骤1:合成化合物7a(Compound 7a)
3,5-二氨基苯甲酸(1.52g,10mmole)和二叔丁氧基二碳酸酯((t-Boc)2O;6.55g,30mmole)与三乙胺(NEt3;7.0mL,60mmole)在CH2Cl2(200mL)中,于室温(25℃)搅拌过夜(24小时)。将得到的溶液用水萃取,用Na2SO4干燥以得到褐色粉末的化合物7a(N-Boc-3,4-二氨基苯甲酸)(2.0g,产率79%)。
化合物7a的化学式:C17H24N2O6。1H NMR(500MHz,d-MeOH)δ7.96(s,1H),7.75(dd,1H,J=8.4,1.9Hz),7.60(d,1H,J=8.4,1.9Hz),4.80(brs,2H,NH),1.52(s,18H).13C NMR(150MHz,d-MeOH)δ173.2,156.4,155.5,135.7,133.6,130.3,127.9,127.8,123.6,81.7,81.6,28.8,28.7。测量化合物7a(C17H24N2O6;352.4Da)的质量。
步骤2:合成化合物7b(Compound 7b)
于化合物7a(500mg,1.42mmole)和五氟苯酚(313mg;1.70mmole)在DCM(8mL)中的混合物溶液添加DCC(351mg,1.70mmole)。接着将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。向PFP酯在DCM(1mL)和DMF(2mL)中的溶液中添加L1(534.8mg,2.12mmole)和DIPEA(457.8mg,3.54mmole)。反应在室温下放置2小时。除去溶剂后,通过制备型HPLC纯化化合物7b(50%CH3CN的H2O溶液+0.1%TFA;UV 210nm;Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物7b,停留时间14.6min)。获得白色固体化合物7b(557mg;产率67%)。
化合物7b的化学式:C27H44N3O11。ESI(阳离子)M/Z:586.2972for C27H44N3O11,err-0.3ppm。
步骤3:合成化合物7c(Compound 7c)
于化合物7b(40mg,68.3mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和Val-Cit-APEA-AF(95mg,85.4mmole)在2mL DMF中的溶液添加TBTU(68mg)和DIPEA(52mg)。3小时后反应完成。随后减压除去溶剂。通过制备型HPLC纯化化合物7c。将含目标化合物的部分冷冻干燥以获得白色固体化合物7c(63.5mg;产率55%)。
化合物7c的化学式:C86H138N14O20。ESI,阳离子:M/Z:1689.2(M+1),1711.2(M+Na)。
步骤4:合成化合物7(Compound 7)
在25~27℃于化合物7c(20.1mg,0.012mmole)在1.5mL DCM中的溶液滴加0.8mL(10.4mmole)的三氟乙酸。接着除去水浴并继续搅拌4小时。随后减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物7c-deBoc 23mg做为TFA盐。
化合物7c-deBoc的化学式:C76H122N14O16。ESI,阳离子:M/Z:1489.0(M+1),1511.0(M+Na),ESI,阴离子:M/Z:1487.2(M-1)。
接下来,在0~5℃搅拌下,于TFA盐(22mg,0.012mmole)的DCM/DMF(1/0.3mL)溶液添加三甲胺(8μL,0.057mmole),随后加入氯乙酰氯化物22mg(0.19mmole)的DCM(0.3mL)溶液。搅拌5分钟后,加入第二部分三甲胺(8μL,0.057mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以2mL M.P.溶剂稀释,Inertsil ODS-3管柱20x250mm,5μm;流速17.5mL/min,M.P含有0.1%TFA的43%AcN/H2O,UV 215nm)。将含产物的部分(Rt=6.5min)冷冻干燥以得到化合物7,即连接子药物7(16mg,产率84%)。
连接子药物7的化学式:C80H124Cl2N14O18。(ESI,阳离子):M/Z:M+H=1641.0。
实施例8:连接子药物8的制备
连接子药物8系根据以下方案所示的步骤合成。
步骤1~3:合成化合物8a~8c(Compounds 8a~8c)
通过与实施例7中所述步骤1~3相同的合成步骤合成化合物8a、8b、和8c。
步骤4:合成化合物8(Compound 8)
在8~10℃于化合物8c(20mg,0.031mmole)在1.5mL DCM中的溶液滴加1mL(13mmole)的三氟乙酸。接着除去水浴并继续搅拌4小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物8c-deBoc 18mg做为TFA盐
接下来,在0~5℃搅拌下,于TFA盐的DCM(1mL)溶液添加三甲胺(13μL,0.093mmole),随后加入溴乙酰溴化物13mg(0.064mmole)的DCM(0.3mL)溶液。搅拌15分钟后,加入第二部分三甲胺(13μL,0.094mmole)。2小时后移除冷却浴,继续搅拌4小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm;流速31mL/min,含有0.1%TFA的44%AcN/H2O,UV 220nm)。将含产物的部分(Rt=7.1min)冷冻干燥以得到化合物8,即连接子药物8(12.4mg)。
连接子药物8的化学式:C80H124Br2N14O18。(ESI,阳离子):M/Z:M+H=1728.3。
实施例9:连接子药物9的制备
连接子药物9系根据以下方案所示的步骤合成。
步骤1:合成化合物9a(Compound 9a)
如文献(Synthesis,1986,48)所述进行化合物9a的合成。3,5-二氨基苯甲酸(0.45g,2.96mmole)和二叔丁氧基二碳酸酯((t-Boc)2O;0.57g,8.9mmole)与三乙胺(NEt3;2.1mL,10.6mmole)在DCM(60mL)中,于室温(25℃)搅拌过夜(24小时)。将得到的溶液用水萃取,用Na2SO4干燥以得到褐色粉末的化合物9a(N-Boc-3,4-二氨基苯甲酸)(0.87g,产率83%)。
化合物9a的化学式:C17H24N2O6。1H NMR(500MHz,CDCl3)δ7.87(s,1H,ArH),7.70(s,2H,ArH),6.90(bs,2H,NH),1.52(s,18H,CH3)。
步骤2:合成化合物9b(Compound 9b)
于化合物9a(74.6mg,0.21mmole)和五氟苯酚(46mg;0.25mmole)在DCM(6mL)中的混合物溶液添加DCC(42mg,0.25mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。接着,于化合物9a在DCM(1mL)和DMF(2mL)中的溶液中添加L1(80mg,0.31mmole)和DIPEA(68mg,0.53mmole)。反应在室温下放置2小时。除去溶剂后,通过制备型HPLC纯化化合物9b(50%CH3CN的H2O溶液+0.1%TFA;UV 210nm;Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物9b,停留时间14.6min)。获得白色固体化合物9b(25mg)。
化合物9b的化学式:C27H43N3O11。ESI(阳离子)M/Z:586.9,608.8。产率:99%。
步骤3:合成化合物9c(Compound 9c)
于化合物9b(25mg,0.04mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和Val-Cit-APEA-AF(48mg,0.04mmole)在2mL DMF中的溶液添加TBTU(44mg)和DIPEA(36mg)。4小时后反应完成。减压除去溶剂。通过制备型HPLC纯化化合物9c(50%CH3CN的H2O溶液+0.1%TFA;UV 210nm;Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物9b,停留时间14.6min)。获得白色固体化合物9c(52mg;产率70%)。
化合物9c的化学式:C86H138N14NaO10。(ESI,阳离子):M/Z:1689.9(M+1).ESI-HIMS(阳离子)M/Z:1710.0214[M+Na]+for C86H138N14NaO10,err:6.4ppm。
步骤4:合成化合物9(Compound 9)
在8~10℃于化合物9c(52mg,0.031mmole)在1.5mL DCM中的溶液滴加0.8mL(10.4mmole)的三氟乙酸。除去水浴并继续搅拌4小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物9c-deBoc 23mg做为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐(35mg,0.021mmole)的DCM(1mL)溶液添加三甲胺(13μL,0.093mmole),随后加入溴乙酰溴化物19mg(0.094mmole)的DCM(0.3mL)溶液。搅拌15分钟后,加入第二部分三甲胺(13μL,0.094mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm;流速32mL/min,含有0.1%TFA的43%AcN/H2O,UV 220nm)。将含产物的部分(Rt=7.6min)冷冻干燥以得到化合物9,即连接子药物9(13.3mg)。
连接子药物9的化学式:C80H124Br2N14O18。(ESI,阳离子):M/Z:M+H=1728.5,ESI-HIMS(阳离子)M/Z:1727.7682,[M+H]+for C60H94Br2N8NaO15,1727.7658err:1.4ppm。
合成Val-Cit-APEA-AF
在实施例9的步骤3中,根据以下方案所示的步骤合成化合物Val-Cit-APEA-AF。
步骤3-1:合成Z-Val-Cit-APEA-Boc
将Z-L-Val-Cit-OH(4.0g,9.79mmole)装入二氯甲烷(250mL)和异丙醇(50mL)的混合物中。然后,将4-氨基苯乙基氨基甲酸叔丁酯(2.95g,12.5mmole)和EEDQ(4.95g,20mmole)加入到混合物中。将混合物在室温(25℃)下搅拌36小时。在40℃下减压除去溶剂,然后将乙醚(300mL)加入到残余物中。将混合物离心1小时。清除醚溶液。将固体产物重新悬浮于乙醚(300mL)中并用超音波处理10分钟。将混合物再次离心1小时。然后,收集固体产物。再次重复这个过程。最后,将收集的固体产物真空干燥,得到4.5g纯度约为90%的目标产物。(HPLC,210nm),产率65.5%。
步骤3-2:合成Z-Val-Cit-APEA-AF
于Z-Val-Cit-APEA-Boc(169mg,0.642mmole)的DCM(4mL)溶液中加入TFA(2mL)。将混合物在室温下搅拌1小时,此后在真空下除去溶剂。粗产物不经进一步纯化而使用。将DeBoc粗化合物Z-Val-Cit-APEA(184mg,0.28mmole)溶解于10mL混合溶剂(DCM:DMF=1:0.1)和AF(300mg,0.35mmole)、TBTU(200mg)和DIPEA(162mg,0.353mmole)。将反应混合物在室温下搅拌17小时。将溶剂真空蒸发并使用制备性管柱进行纯化(50%CH3CN的H2O溶液+0.1%TFA;UV 210nm;Inertsil ODS-3管柱30x250mm;流速32mL/min;Z-Val-Cit-APEA-AF,停留时间14.6min,223mg,产率62%)。
Z-Val-Cit-APEA-AF的化学式:C67H103N11O12。ESI(阳离子)M/Z:1256.1。
步骤3-3:合成Val-Cit-APEA-AF
于Z-Val-Cit-APEA-AF(90mg,0.0717mmole)的乙醇(10mL)溶液中加入10%Pd/C(9mg)。将所得混合物在H2气氛下搅拌4小时,此后将混合物通过硅藻土垫过滤。硅藻土垫用EtOAc洗涤,滤液真空浓缩,得到产物Val-Cit-APEA-AF。产率:99%。
Val-Cit-APEA-AF的化学式:C59H97N11O10。ESI-HIMS(阴离子)M/Z:1142.7316[M+Na]+for C59H97N11NaO10,err:-0.3ppm。
实施例10:连接子药物10的制备
连接子药物10系根据以下方案所示的步骤合成。
步骤1:合成化合物10a(Compound 10a)
将二胺(1当量)的DCM溶液(10mL/mmole)用Boc2O(0.15当量)在室温下处理5小时。有机相用水洗涤,直至提取所有未反应的二胺。Boc-保护的化合物10a在干燥(MgSO4)并真空浓缩后定量回收。1H NMR(CDC13,500MHz)3.74(m,1H),3.70(m,1H),3.62(m,8H),3.54(m,4H),3.31(m,2H),1.44(s,9H),ESI,阳离子:M/Z:293.7(M+1),316.7(M+Na)。
步骤2:合成化合物10b(Compound 10b)
于澳瑞他汀F(200mg,0.23mmole)在4mL共溶剂DCM/DMF(1:1)的溶液和化合物10a(75mg,0.30mmole)在2mL DMF的溶液添加TBTU(220mg,0.68mmole)和DIPEA(180mg,1.39mmole)。反应于2小时后结束。接着减压除去溶剂。通过制备性HPLC进行纯化(Inertsil ODS-3管柱30x250mm,10μm;乙腈43%,流速32mL/min,UV220,停留时间9.3min)。获得白色固体化合物10b(151mg,0.14mmole,64%)。ESI,阳离子:M/Z:1021.7(M+1),1043.7(M+Na)。
步骤3:合成化合物10c(Compound 10c)
于化合物10b(151mg)的DCM(4mL)溶液中添加TFA(2mL)。将反应混合物在室温搅拌1小时,此后在真空下除去溶剂。化合物10c的粗产物不经进一步纯化而使用。
步骤4:合成化合物10d(Compound 10d)
于N-Boc-3,4-二氨基苯甲酸(60mg,0.17mmole)和五氟苯酚(47mg;0.25mmol)在DCM(6mL)中的混合物溶液添加DCC(51.5mg,0.25mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。向PFP酯溶液添加化合物10c(235mg,0.25mmole)和DIPEA(55mg,0.42mmole)。反应在室温下放置1小时。除去溶剂后,通过制备型HPLC纯化化合物10d(Inertsil ODS-3管柱30x250mm,5μm,乙腈42%(0~5min)至50%(5~13min)至55%(13~25min),流速31mL/min,UV220,室温13.8min)。获得白色固体化合物10d(173mg,0.13mmole,81%)。ESI,阳离子:M/Z:1255.3,12773(M+Na)。
步骤5:合成化合物10(Compound 10)
在20~23℃于化合物10d(20mg,0.016mmole)在4mL DCM中的溶液滴加2mL(10.4mmole)的三氟乙酸。除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物10d-deBoc 23mg做为TFA盐。
在0~5℃搅拌下,于TFA盐(23mg,0.016mmole)的DCM/DMP(1/0.5mL)溶液添加三甲胺(10μL,0.072mmole),随后加入溴乙酰溴化物24mg(0.12mmole)的DCM(0.3mL)溶液。搅拌5分钟后,加入第二部分三甲胺(10μL,0.072mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以2mL M.P.溶剂稀释,Inertsil ODS-3管柱20x250mm,5μm;流速17.5mL/min,M.P.含有0.1%TFA的42%AcN/H2O,UV215nm)。将含产物的部分(Rt=7.47min)冷冻干燥以得到化合物10,即连接子药物10(12mg,产率58%)。
连接子药物10的化学式:C59H93Br2N9O13。(ESI,阳离子):M/Z:M+H=1297.5。
实施例11:连接子药物11的制备
连接子药物11系根据以下方案所示的步骤合成。
步骤1:合成化合物11a(Compound 11a)
将L1(0.79g,3.14mmole)和K2CO3(1.04g,7.50mmole)溶于5.8mL水中,并在室温下搅拌15分钟。之后,加入N-(9-芴基甲氧基羰基)琥珀酰亚胺(Fmoc-OSu,1.28g,3.79mmole)并将混合物搅拌24小时。通过TLC(CHCl3/MeOH/AcOH=5:1:0.06)监测反应的过程。过滤盐并将滤液用Et2O洗涤,使用3N HCl酸化至pH 1,并将所需化合物用DCM萃取。以真空除去溶剂后,通过RP-HPLC纯化,将含有产物的部分冷冻干燥,得到化合物11a(1.08g,产率73%)。
化合物11a的化学式:C25H31NO8。所需[M+H]+=474.2。所得[M+H]+=474.9,[M+Na]+=496.9。
步骤2:合成化合物11b(Compound 11b)
在室温下将咪唑(0.78g,11.5mmole)悬浮于10mL DCM中。分批加入Boc2O(2.62g,12mmole)。将反应混合物在室温下搅拌1小时。反应混合物用水洗涤,用Na2SO4干燥,过滤,减压除去挥发物。将残余物溶于4mL甲苯中并加入二亚乙基三胺(0.595mL,5.5mmole)。将反应混合物在60℃下搅拌2小时。加入10mL DCM,并将有机相用水洗涤。有机相用Na2SO4干燥,过滤并减压下还原。在快速管柱二氧化硅上使用梯度的具有三乙胺在DCM中的甲醇(MeOH),得到目标化合物,为无色固体(化合物11b,1.02g,产率61%)。
化合物11b的化学式:C14H29N3O4。1H-NMR(500MHz,CDCl3):1.41(s,18H),1.58(bs,1H),2.66-2.77(m,4H),3.13-3.26(m,4H),4.96(bs,2H)。
步骤3:合成化合物11c(Compound 11c)
于化合物11a(645.2mg,1.36mmole)在4mL DCM的溶液和化合物11b(413mg,1.36mmole)在2mL DMF的溶液添加TBTU(110mg)和DIPEA(844mg)。反应于2小时后结束。减压除去溶剂。通过RP-HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物11c,停留时间17min)。获得白色固体化合物11c(738mg;yield 76%)。
化合物11c的化学式:C39H58N4O11Na。(ESI,阳离子):M/Z:760.4(M+1);782.4(M+Na)。所得HRMS 781.4005for C39H58N4O11Na,err:1.3ppm。
步骤4:合成化合物11d(Compound 11d)
在0℃于化合物11c(218.6mg,0.288mmole)在4mL DCM中的溶液滴加2mL的三氟乙酸。除去水浴并继续搅拌3小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物11c-deBoc 27mg做为TFA盐。
化合物11c-deBoc的化学式:C29H42N4O7。ESI,阳离子):M/Z:558.4(M+1);580.3(M+Na)。
向TFA盐(250mg,0.44mmole)在4mL DCM中的溶液和化合物N-Boc-L1(628mg,1.76mmole)在2mL DMF中的溶液添加TBTU(831.6mg)和DIPEA(552mg)。3小时后反应完成。减压除去溶剂。通过制备型HPLC纯化化合物11d。将含目标化合物的部分冷冻干燥以获得白色固体化合物11d(356mg;产率65%)。
化合物11d的化学式:C59H96N6O21。ESI,阳离子:M/Z:1226.7(M+1),1248.8(M+Na)。
步骤5:合成化合物11e(Compound 11e)
在0℃于化合物11d(300mg,0.288mmole)在4mL DCM中的溶液滴加2mL的三氟乙酸。接着除去水浴并继续搅拌3小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物11d-deBoc做为TFA盐。
化合物11d-deBoc的化学式:C49H80N6O17。所需[MH+]1024.56,所得[M+H]+1026.7.[M+Na]+1048.7。
接下来,于TFA盐(130mg,0.12mmole)在2mL DCM中的溶液和澳瑞他汀F(272mg,0.3mmole)在2mL DMF中的溶液添加TBTU(235mg,0.73mmole)和DIPEA(173mg,1.34mmole)。1小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流速32mL/min;化合物11e,停留时间17min)。获得白色固体化合物11e(261mg;产率83%)。
化合物11e的化学式:C129H210N16O31。(ESI,阳离子):M/Z:1242.3(M+2H)2+;829.0(M+3H)3+。
步骤6:合成化合物11f(Compound 11f)
在0℃于化合物11e(20.8mg,0.288mmole)在2mL DCM中的溶液中逐滴加入1mL二乙胺。移除冷却浴,继续搅拌4小时。接着减压除去溶剂。于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物11e-deFmoc做为TFA盐。
化合物11e-deFmoc的化学式:C114H200N16O29。ESI,阳离子:2259.5(M+1),2281.5(M+Na)。
接下来,于CJ24-1(64mg,0.11mmole)在4mL DCM中的溶液和化合物11e-deFmoc(123.5mg,0.054mmole)在2mL DCM中的溶液添加EDC(26mg,0.13mmole)和DIPEA(14mg,0.11mmole)。12小时后反应完成。减压除去溶剂。通过制备型HPLC纯化化合物11f。将含目标化合物的部分冷冻干燥以获得白色固体化合物11f(169.8mg;产率55%)。
化合物11f的化学式:C125H211N19O33。ESI,阳离子:1415(M+2H)2+。
步骤7:合成化合物11(Compound 11)
在20~23℃于化合物11f(26.3mg,0.0092mmole)在4mL DCM中的溶液滴加2mL(10.4mmole)的三氟乙酸。除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物11f-deBoc 26mg做为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐(26mg,0.0092mmole)的DCM/DMAc(1/0.5mL)溶液添加三甲胺(40μL,0.288mmole)的DCM(0.2mL)溶液,随后加入溴乙酰溴化物30mg(0.148mmole)的DCM(0.3mL)溶液。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3.5mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.从35%(0-6.5min)至43%(6.5-20min)含有0.1%TFA的AcN/H2O,UV220nm)。将含产物的部分(Rt=11.71min)冷冻干燥以得到化合物11,即连接子药物11(12mg,产率46%)。
连接子药物11的化学式:C135H227Br2N19O37。(ESI,阳离子):M/Z:[M+H]+2=1435.9。
实施例12:连接子药物12的制备
连接子药物12系根据以下方案所示的步骤合成。
步骤1:合成化合物12a(Compound 12a)
将二叔丁基碳酸酯(5.2g,23.8mm)的乙腈溶液缓慢加入经搅拌的(S)-5-(氨基甲基)吡咯烷-2-酮(EDA-4,1.29g,11.3mmole),接着加入DMAP(180mg,1.5mmole)。反应于6小时后结束(以TLC监测)。将溶液浓缩,通过快速管柱层析法(己烷/EtoAC(1:5))纯化固体,得到固体状的二-Boc-保护的中间体EDA-5(1.82g,51%)。将LiOH×H2O(0.36g,19mmole)在水(12ml)中的溶液加入到EDA-5(1.03g,3.28mmole)的10ml THF中。将混合物搅拌过夜,然后用1N HCl酸化。将溶液浓缩,并将混合物用EtOAc萃取,用盐水洗涤,用MgSO4干燥并浓缩。通过快速管柱层析法(乙酸乙酯/EtoAC(1/4))纯化固体,得到固体状的化合物12a(0.81g,75%)。M/Z=333.9[M+H]+,M/Z=331.8[M-1]-;.1H NMR(CDCl3)1.41(s,9H);1.43(s,9H);1.61-1.84(m,2H),2.42-2.45(m,2H),3.17-3.22(t,2H),3.67(s,1H),4.95(bs,2H)。
步骤2:合成化合物12b(Compound 12b)
于化合物12a(133mg,0.40mmole)和五氟苯酚(110mg;0.60mmol)在DCM(6mL)中的混合物溶液添加DCC(123mg,0.60mmole)。接着将混合物在室温下搅拌12小时,得到粗产物化合物12b。
步骤3:合成化合物12c(Compound 12c)
加入CJ35-5(151mg,0.60mmole)和DIPEA(130mg,1.00mmole)。反应在室温下放置2小时。除去溶剂后,通过制备型HPLC纯化化合物12c(50%CH3CN的H2O溶液+0.1%TFA;UV 220nm;Inertsil ODS-3管柱30x250mm;流速31mL/min;化合物12c,停留时间6.9min)。获得白色固体化合物12c(160mg;产率70%)。MS(ESI,阴离子):M/Z:564.8(M-H)+。
步骤4:合成化合物12d(Compound 12d)
于化合物12c(60mg,0.106mmole)在4mL共溶剂DCM/DMF(2:2)中的溶液和Val-Cit-APEA-AF(118.8mg,0.106mmole)在2mL DMF中的溶液添加TBTU(113mg,0.297mmole)和DIPEA(79.5mg,0.616mmole)。4小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;10μm;乙腈43%,流速32mL/min,UV220,化合物12d,停留时间9.3min)。获得白色固体化合物12d(113.8mg,0.068mmole;产率64.2%)。ESI,阳离子:M/Z:1671.7(M+1),1693.7(M+Na)。
步骤5:合成化合物12(Compound 12)
在8~10℃于化合物12d(24mg,0.014mmole)在1.5mL DCM中的溶液滴加三氟乙酸(1mL,13mmole)。接着除去冰浴并继续搅拌4小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物12d-deBoc 19mg做为TFA盐。
接下来,在0~5℃搅拌下,于TFA盐的DCM(1mL)溶液添加三甲胺(13μL,0.093mmole),随后加入溴乙酰溴化物22mg(0.119mmole)的DCM(0.3mL)溶液。搅拌15分钟后,加入第二部分三甲胺(13μL,0.094mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3mL移动相溶剂稀释,Inertsil ODS-3管柱30x250mm;流速31mL/min,44%can/H2O(0~15min),50%(15~25min),化合物12,即连接子药物12,18.3min,UV 220nm)。将含产物的部分(Rt=18.3min)冷冻干燥以得到化合物12,即连接子药物12(1.4mg)。(ESI,阳离子):M/Z:M+H=1711.8(Br79,M+1),1713.9(Br81,M+1)。
实施例13:连接子药物13的制备
连接子药物13系根据以下方案所示的步骤合成。
步骤1:合成化合物13a(Compound 13a)(1,3-二(叔丁氧基羰基氨基)丙-2-醇)
根据文献方法(Chem.Eur.J.2004,10,1215-1226)制备化合物13a。
将二叔丁氧基二碳酸酯(4.80g,22mmol)的CH2Cl2(4mL)溶液缓慢加入经搅拌的1,3-二氨基丙醇(901mg,10.0mmol)和三乙胺(164mL,1.18mmol)的THF/MeOH(1:5,10mL)中。反应于2小时后结束(以TLC监测)。将溶液浓缩,通过快速管柱层析法(己烷/EtoAC(1:4))纯化油性残留物,得到无色固体状的Boc-保护的中间体化合物13a(1.59g,55%)。1H NMR(500MHz,CDCl3)δ=5.16(bs,2H),3.73(m,1H),3.22-3.16(m,4H),1.43(s,18H)ppm;M/Z(ES+),291.7(M+H),313.7(M+Na)。
步骤2:合成化合物13b(Compound 13b)(N-(3-(叔丁氧基羰基氨基)-2-(乙氧基羰基甲氧基)-丙-1基)氨基甲酸叔丁酯)
在室温下,将溴乙酸叔丁酯(2.07g,10.7mmol)加入到经搅拌化合物13a(1.23g,4.23mmol)在无水THF(2mL)中的溶液。在1小时内缓慢加入氢化钠(0.47g,4.5摩尔当量)。再过5小时后,将反应混合物用硅藻土过滤并蒸发。通过快速管柱层析法(己烷/EtOAc(8/2))纯化残留物,得到无色油状的化合物13b。1H NMR(500MHz,CDCl3):=4.21(q,J=7Hz,2H),4.15(s,2H),3.45(m,1H),3.48-3.06(m,4H),1.42(s,18H),1.27(t,J=7Hz,3H);MS m/z(ES-),348.2(M-t-BuO)。
步骤3:合成化合物13c(Compound 13c)(N-(3-(叔丁氧基羰基氨基)-2-(乙氧基羰基甲氧基)-丙-1基)氨基甲酸)
将化合物13b(30mmol)溶于THF(100mL)和MeOH(100mL)中,加入6N NaOH(150mL),将反应混合物在室温下搅拌1小时。在真空下除去溶剂,并在0℃加入6N HCl(155mL)。用CH2Cl2萃取后,用Na2SO4干燥,滤去干燥剂,蒸去溶剂,得粗产物。通过管柱层析法进行纯化(硅胶,己烷/乙酸乙酯=3/1至1/3)已获得白色固体(3.9g,74.5%)。1H NMR(500MHz,CDCl3):δ5.21(br,1H),4.72(br,1H),4.62(br,1H),3.92(s,2H),3.71-3.68(t,1H),3.44-3.29(m,4H),1.39(s,18H,CH3);MS m/z(ES-),347.7(M-1),273.6(M-t-BuO)。
步骤4:合成化合物13d(Compound 13d)
于化合物DETA-4(58mg,0.16mmole)和五氟苯酚(36.8mg;0.20mmol)在DCM(6mL)中的混合物溶液添加DCC(41.2mg,0.20mmole)。将混合物在室温下搅拌12小时。加入CJ35-5(62.7mg,0.25mmole)和DIPEA(54mg,0.41mmole)。反应在室温下放置3小时。除去溶剂后,通过制备型HPLC纯化化合物1c(50%至35%CH3CN的H2O溶液+0.1%TFA;UV 220nm;Inertsil ODS-3管柱30x250mm;流速31mL/min;化合物13d)。获得白色固体化合物13d(73mg;产率73%)。MS(ESI,阴离子):M/Z:581.1(M-H)+。
步骤5:合成化合物13e(Compound 13e)
于化合物13d(57.5mg,0.098mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和MMAE(70.5mg,0.098mmole)在2mL DMF中的溶液添加TBTU(47.6mg,0.14mmole)和DIPEA(31.8mg,0.24mmole)。4小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;10μm;流速32mL/min;UV 220,化合物13e)。获得白色固体化合物13e(101.6mg;产率80.2%)。ESI,阳离子:M/Z:1286.9(M+1),1304.9(M+Na)。
步骤6:合成化合物13(Compound 13)
在8~10℃于化合物13e(12mg,0.009mmole)在1mL DCM中的溶液滴加0.8mL(10.4mmole)的三氟乙酸。除去冰水浴并于室温继续搅拌4小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物13e-deBoc 11mg做为TFA盐。
在0~3℃搅拌下,向TFA盐的DCM(1mL)溶液添加三甲胺(20μL,0.136mmole),随后加入溴乙酰溴化物15mg(0.074mmole)的DCM(0.25mL)溶液。搅拌15分钟后,加入第二部分三甲胺(10μL,0.068mmole)。2小时后移除冷却浴,继续搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm;10μm;乙腈43%(0~9min)50%(9~15min)60%(15~25min);流速31mL/min;UV 220nm)。将含产物的部分(Rt=14.1min)冷冻干燥以得到化合物13,即连接子药物13(3.5mg)。(ESI,阳离子):M/Z:M+H=1322.7(Br79,M+1)1324.7(Br81,M+1)。
实施例14:连接子药物14的制备
连接子药物14系根据以下方案所示的步骤合成。
步骤1:合成化合物14a(Compound 14a)
将10g的2,2’-(亚乙二氧基)双(乙基胺)的DCM溶液(100mL)用Boc2O(2.25g,0.01mole)在0℃下处理5小时,在室温下处理18小时。有机相用100mL的水洗涤,直至提取所有未反应的二胺。Boc-保护的化合物10a在干燥(MgSO4)并真空浓缩后定量回收(2.53g,0.01mole)。1H NMR(500MHz,CDCl3):d 1.42(s,9H),1.63(br m,2H,NH2),2.86(t,2H,J=5.5Hz),3.30(m,2H),3.50(m,4H),3.49-3.61(m,8H),5.30(br s,1H,NHCO2)。
步骤2:合成脯氨酸-AF
于澳瑞他汀F(500mg,0.58mmole)在12mL DCM和脯氨酸叔丁酯(110mg,0.64mmole)添加HATU(330mg,0.86mmole)和DIPEA(188mg,1.45mmole)。反应于3~4小时后结束。接着减压除去溶剂。向上述的DCM(6mL)溶液中加入TFA(2mL)。将混合物在室温下搅拌5小时,然后真空除去溶剂。通过制备性HPLC进行纯化(Inertsil ODS-3管柱30x250mm,流速31mL/min,流动相:乙腈35%(0~7min)43%(7-16min)43%(7-16min)100%(16min-),具有0.1%TFA的水,UV 220nm,脯氨酸-AF,停留时间9.3min)。获得白色固体脯氨酸-AF(441.2mg,90%)。ESI(阳离子),844.1[M+H]+,866.6[M+Na]+。
步骤3:合成化合物14b(Compound 14b)
于脯氨酸-AF(504.3mg,0.59mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和化合物N-Boc-PEG-胺(N-Boc-2,2’-(ethylenedioxy)diethylamine),163mg,0.106mmole)在2mL DMF中的溶液添加HATU(340mg,0.89mmole)和DIPEA(193mg,1.49mmole)。2小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;10μm;乙腈43%(0~15min)100%(15min~);流速32mL/min;UV 220nm,化合物14b,停留时间11.5min)。获得白色固体化合物14b(530mg,产率82.8%)。ESI,阳离子:M/Z:1074.6[M+H]+,1096.9[M+Na]+。
步骤4:合成化合物14c(Compound 14c)
于化合物14b(530mg)的DCM(4mL)溶液添加TFA(2mL)。将混合物在0℃下搅拌1.6小时,然后真空除去溶剂。化合物14c的粗产物(TFA盐)不经进一步纯化而使用。ESI,阳离子:M/Z:974.8[M+H]+,996.9[M+Na]+。
步骤5:合成化合物14d(Compound 14d)
于N-Boc-3,4-二氨基苯甲酸(1g,2.84mmole)和五氟苯酚(784mg;4.26mmol)在DCM(18mL)中的混合物溶液添加DCC(880mg,4.27mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。以正己烷/EtOAc(4:1)洗脱管柱。收集含目标化合物的部分,减压浓缩以得到N-Boc-3,4-二氨基苯甲酸PFP酯(1.47g,2.84mmole,产率80%)。1H-NMR(500MHz;CDCl3)d:8.12(s;1H);7.97(s;2H),7.25(s;1H),1.5(s,18H),ESI,阴离子):M/Z:517.9(M-)。
于N-Boc-3,4-二氨基苯甲酸PFP酯溶液(255mg,0.49mmole)在4mL共溶剂DCM/DMF(1:1)中的溶液和化合物14c(480.6mg,0.49mmole)在2mL DMF中的溶液添加DIPEA(317.5mg,2.46mmole)。5小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;10μm,乙腈35%(0~5min)43%(5~11min)50%(11~20min)55%(20~25min)65%(25~30min)100%(30min~);流速32mL/min,UV220nm,化合物14d,停留时间13.8min)。获得白色固体14d(450mg,0.344mmole,产率70%)。ESI(阳离子):1309.0[M+H]+,1331.0[M+Na]+。
步骤6:合成化合物14(Compound 14)
在20~23℃于化合物14d(25mg,19μmole)在4mL DCM中的溶液滴加2mL(10.4mmole)的三氟乙酸。除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物14d-deBoc做为TFA盐。ESI(阳离子):1108.9[M+H]+,1130.9[M+Na]+。
在0~5℃搅拌下,于TFA盐(24mg,19μmole)的DCM/DMF(1.2/0.5mL)溶液添加三甲胺(40μL,0.288mmole)的DCM(0.2mL)溶液,随后加入氯乙酰氯化物21mg(0.288mmole)的DCM(0.3mL)溶液。2小时后移除冷却浴,继续于室温搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3.5mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.具有0.1%TFA的乙腈35%(0~5.5min)43%(5.5~16min)100%(16min~),UV 220nm)。将含产物的部分(Rt=10.81min)冷冻干燥以得到化合物14,即连接子药物14(20mg,产率83%)。ESI,阳离子:M/Z:1260.8[M+H]+,1282.9[M+Na]+=1282.9。
实施例15:连接子药物15的制备
连接子药物15系根据以下方案所示的步骤合成。
步骤1:合成化合物15a(Compound 15a)
于脯氨酸-AF(51.3mg,0.06mmole)在6mL DCM的溶液和化合物N-Boc-1,6-己二胺(N-Boc-1,6-hexanediamine,CAS:194920-62-2),22.4mg,0.07mmole)在2mL DCM中的溶液添加HATU(34.2mg,0.09mmole)和DIPEA(19.3mg,0.15mmole)。4小时后反应完成。接着减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流速31mL/min;流动相从乙腈35%(0~5min)43%(5~21min)100%(21min~),0.1%TFA;UV 220nm,化合物15a,停留时间13.6min)。获得白色固体化合物15a(52mg,产率74.6%)。ESI,阳离子:M/Z:1146.7[M+1]+,1169.0[M+Na]+。
步骤2:合成化合物15b(Compound 15b)
于化合物15a(52mg,0.05mmole)的DCM(4mL)溶液中添加TFA(2mL)。将反应混合物在室温搅拌1小时,此后在真空下除去溶剂。化合物15b的粗产物不经进一步纯化而使用。ESI,阳离子:M/Z:1046.9[M+1]+,1069.0[M+Na]+。
步骤3:合成化合物15c(Compound 15c)
于N-Boc-3,4-二氨基苯甲酸(16mg,0.04mmole)和五氟苯酚(12.5mg;0.06mmol)在DCM(18mL)中的混合物溶液添加DCC(12.3mg,0.06mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。以正己烷/EtOAc(4:1)洗脱管柱。收集含目标化合物的部分,减压浓缩以得到N-Boc-3,4-二氨基苯甲酸PFP酯。
于N-Boc-3,4-二氨基苯甲酸PFP酯溶液添加化合物15b(47.4mg,0.03mmole)的2mL DMF溶液和DIPEA(29.3mg,0.23mmole)。2小时后反应完成。减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;10μm,具有0.1%TFA的乙腈43%(0~5min)50%(5~10min)55%(10~15min)65%(15~20min)100%(20min~);流速32mL/min,UV220nm,停留时间13.9min)。获得白色固体15c(28.9mg,0.022mmole,产率50%)。ESI(阳离子):1380.9[M+1]+,1403.2[M+Na]+。
步骤4:合成化合物15(Compound 15)
在20~23℃于化合物15c(8.6mg,6μmole)在4mL DCM中的溶液滴加2mL(10.4mmole)的三氟乙酸。除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物15c-deBoc(8.1mg)做为TFA盐。ESI(阳离子):1277.0[M+1]+,1298.9[M+Na]+。
在0~5℃搅拌下,于TFA盐(8.1mg,6μmole)的DCM/DMF(1.2/0.5mL)溶液添加三甲胺(45μL,0.324mole)的DCM(0.2mL)溶液,随后加入氯乙酰氯化物21mg(0.185mmole)的DCM(0.3mL)溶液。2小时后移除冷却浴,继续于室温搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以4mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.具有0.1%TFA的乙腈35%(0~5min)43%(5~15min)100%(15min~),UV 220nm)。将含产物的部分(Rt=11.45min)冷冻干燥以得到化合物15,即连接子药物15(6mg,4.5μmole,产率75%)。ESI,阳离子:M/Z:1333.0[M+H]+,1355.0[M+Na]+。
实施例16:连接子药物16的制备
连接子药物16系根据以下方案所示的步骤合成。
步骤1:合成化合物16a(Compound 16a)
于脯氨酸-AF(117mg,0.14mmole)在8mL DCM的溶液和化合物N-Boc-1,6-己二胺(N-Boc-1,6-hexanediamine,45mg,0.21mmole)添加HATU(80mg,0.21mmole)和DIPEA(45mg,0.35mmole)。1小时后反应完成。接着减压除去溶剂。通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流速31mL/min;流动相从乙腈43%(0~10min)50%(10~17min)100%(17min~),0.1%TFA;UV 220nm,化合物16a,停留时间14.4min)。获得白色固体化合物16a(137.8mg,产率95%)。ESI,阳离子:M/Z:1042.4[M+H]+,1064.9[M+Na]+。
步骤2:合成化合物16b(Compound 16b)
于化合物16a(137.8mg,0.13mmole)的DCM(4mL)溶液中添加TFA(2mL)。将反应混合物在室温搅拌1小时,此后在真空下除去溶剂。化合物16b的粗产物不经进一步纯化而使用。ESI,阳离子:M/Z:942.6[M+H]+,964.8[M+Na]+。
步骤3:合成化合物16c(Compound 16c)
于N-Boc-3,4-二氨基苯甲酸(12.7mg,0.03mmole)和五氟苯酚(10mg;0.05mmol)在DCM(6mL)中的混合物溶液添加DCC(11.1mg,0.05mmole)。将混合物在室温下搅拌12小时,过滤沉淀物并真空浓缩。向PFP酯溶液添加化合物16b(33.9mg,0.03mmole)和DIPEA(27.3mg,0.18mmole)。反应在室温下放置1小时。除去溶剂后,通过制备型HPLC进行纯化(Inertsil ODS-3管柱30x250mm;流速31mL/min,流动相:乙腈43%(0~5min)50%(5~10min)55%(10~20min)100%(20min~);0.1%TFA的水;UV220nm,停留时间15.2min)。获得白色固体16c(32mg,产率70%)。ESI,阳离子:M/Z:1277.0[M+H]+,1299.0[M+Na]+。
步骤4:合成化合物16(Compound 16)
在20~23℃于化合物16c(7.5mg,5.8μmole)在4mL DCM中的溶液滴加2mL(10.4mmole)的三氟乙酸。除去水浴并继续搅拌2小时。减压除去溶剂,于残余物中加入2mL甲醇,搅拌5分钟,接着除去溶剂。加入最少量的DIW(2mL)并冷冻干燥以得到粗产物化合物16c-deBoc做为TFA盐。ESI for阳离子,M/Z:1077.0[M+H]+,1098.9[M+Na]+。
在0~5℃搅拌下,于TFA盐(7mg,5.8μmole)的DCM/DMF(1.0/0.5mL)溶液添加三甲胺(40μL,0.288mmole)的DCM(0.2mL)溶液,随后加入氯乙酰氯化物18mg(0.159mmole)的DCM(0.3mL)溶液。2小时后移除冷却浴,继续于室温搅拌15小时。通过HPLC检查反应是否完成,并将混合物蒸发至干燥。以RP-HPLC进行纯化(残余物以3.5mL M.P.溶剂稀释,Inertsil ODS-3管柱30x250mm,10μm;流速31mL/min,M.P.从35%至43%具有0.1%TFA的乙腈,UV 220nm)。将含产物的部分(Rt=12.27min)冷冻干燥以得到化合物16,即连接子药物16(5mg,4.1μmole,产率70%)。ESI,阳离子:M/Z:[M+H]+=1228.9,1250.9[M+Na]+。
<抗体的还原>
用2.0~5.5摩尔当量的TCEP在37℃下处理2小时进行赫赛汀的还原。脱盐后,用DTNB测定赫赛汀的巯基的浓度,用UV 280nm测定蛋白质浓度,计算SH/IgG的数量。还原的赫赛汀的图谱由CE获得。确定4对链间双硫键可以通过4.5-5.0摩尔当量的TCEP还原以获得8个游离巯基。
<抗体药物复合物(ADC)>
分别使用毛细管电泳(CE)和疏水作用层析法(HIC)测定交联度和平均药物抗体比(DAR)。
疏水作用层析法(Hydrophobic Interaction Chromatography;HIC)分析
使用具有Butyl NPR(4.6×35mm)TOSOH管柱的Agilent HPLC分析药物抗体比例(DAR)图谱。移动相A包括25mM的磷酸钠、1.5M硫酸铵、pH6.95,且移动相B包括25mM磷酸钠、25%异丙醇、pH 6.95。将10μL的样品以0.8mL/min的流速注入管柱,并在梯度模式(gradient mode)下进行分离20分钟(表2)。侦测在波长280nm的吸收度。
表2
实施例17:赫赛汀-连接子药物1
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用0.77μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在25~40℃下将2.618μL(17摩尔当量)、20mM、在DMSO中制备的连接子药物1加入到抗体溶液中2小时(在混合溶液中有机溶剂的最终浓度为约4.9%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物1。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图1显示赫赛汀-连接子药物1的平均DAR为约3.9,D4比率为约65%。
实施例18:赫赛汀-连接子药物2
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。加入2.8μL的DMSO并混合均匀。接着,在0~4℃下将1.386μL(9摩尔当量)、20mM、在DMSO中制备的连接子药物2加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约7.6%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物2。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图2显示赫赛汀-连接子药物2的平均DAR为约4.3,D4比率为约60%。
实施例19:赫赛汀-连接子药物3
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在0~4℃下将3.234μL(21摩尔当量)、20mM、在DMSO中制备的连接子药物3加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约5.9%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物3。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图3显示赫赛汀-连接子药物3的平均DAR为约3.6,D4比率为约31%。
实施例20:赫赛汀-连接子药物5
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在0~4℃下将1.386μL(9摩尔当量)、20mM、在DMSO中制备的连接子药物5加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约2.7%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物5。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图4显示赫赛汀-连接子药物5的平均DAR为约4.0,D4比率为约54%。
实施例21:赫赛汀-连接子药物7
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。加入9.9μL的DMSO并混合均匀。接着,在0~4℃下将1.694μL(11摩尔当量)、20mM、在DMSO中制备的连接子药物7加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约18.3%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物7。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图5显示赫赛汀-连接子药物7的平均DAR为约3.9,D4比率为约77%。
实施例22:赫赛汀-连接子药物8
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用33.87μL的TCEP(5摩尔当量)处理440μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。加入76.1μL的DMSO并混合均匀。接着,在25~40℃下将15.25μL(9摩尔当量)、20mM、在DMSO中制备的连接子药物8加入到抗体溶液中2小时(在混合溶液中有机溶剂的最终浓度为约12.8%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物8。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图6显示赫赛汀-连接子药物8的平均DAR为约4.2,D4比率为约65%。
实施例23:赫赛汀-连接子药物9
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。加入12.5μL的DMSO并混合均匀。接着,在25~40℃下将0.924μL(6摩尔当量)、20mM、在DMSO中制备的连接子药物9加入到抗体溶液中2小时(在混合溶液中有机溶剂的最终浓度为约28.9%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物9。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图7显示赫赛汀-连接子药物9的平均DAR为约4.0,D4比率为约70%。
实施例24:赫赛汀-连接子药物11
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在0~4℃下将1.078μL(7摩尔当量)、20mM、在DMSO中制备的连接子药物11加入到抗体溶液中2小时(在混合溶液中有机溶剂的最终浓度为约2.1%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物11。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图8显示赫赛汀-连接子药物11的平均DAR为约8.0,D8比率为约53%。赫赛汀-连接子药物11的连接子单元接合2个药物单元。因此,赫赛汀-连接子药物11的平均DAR为其他的ADC的两倍。
实施例25:赫赛汀-连接子药物12
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在25~40℃下将2.615μL(17摩尔当量)、20mM、在DMSO中制备的连接子药物12加入到抗体溶液中2小时(在混合溶液中有机溶剂的最终浓度为约4.9%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物12。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图9显示赫赛汀-连接子药物12的平均DAR为约3.2,D4比率为约38%。
实施例26:赫赛汀-连接子药物13
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在25~40℃下将0.924μL(6摩尔当量)、20mM、在DMSO中制备的连接子药物13加入到抗体溶液中2小时(在混合溶液中有机溶剂的最终浓度为约1.8%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物13。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图10显示赫赛汀-连接子药物13的平均DAR为约3.9,D4比率为约58%。
实施例27:赫赛汀-连接子药物14
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用0.77μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在0~4℃下将2.002μL(13摩尔当量)、20mM、在DMSO中制备的连接子药物14加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约3.8%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物14。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图11显示赫赛汀-连接子药物14的平均DAR为约4.0,D4比率为约87%。
实施例28:赫赛汀-连接子药物15
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用0.77μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。在0~4℃下将2.002μL(13摩尔当量)、20mM、在DMSO中制备的连接子药物15加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约3.8%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物15。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图12显示赫赛汀-连接子药物15的平均DAR为约4.0,D4比率为约82%。
实施例29:赫赛汀-连接子药物16
将抗体赫赛汀(购自Roche)的冷冻晶体溶于DIW(初始浓度:11.36mg/mL)中。用3.07μL的TCEP(5摩尔当量)处理40μL的抗体溶液,并在37℃下搅拌2小时。使用脱盐管柱除去还原的赫赛汀中的多余TCEP,并将缓冲溶液更换为硼酸盐缓冲溶液。加入16.0μL的DMSO并混合均匀。接着,在0~4℃下将2.002μL(13摩尔当量)、20mM、在DMSO中制备的连接子药物16加入到抗体溶液中24小时(在混合溶液中有机溶剂的最终浓度为约26.4%)以获得抗体药物复合物(ADC)。使用脱盐柱(ThermoFisher Scientific,MWCO:40K)纯化产物ADC赫赛汀-连接子药物16。在洗脱期间,将缓冲溶液变为PBS缓冲溶液(2.67mM KCl,1.47mM KH2PO4,137.93mM NaCl,8.06mM Na2HPO4·7H2O)。
图13显示赫赛汀-连接子药物16的平均DAR为约4.0,D4比率为约81%。
<储存稳定性测试>
将20mM连接子药物7在-20℃下于DMSO中保存0、21、92、和99天。LC-MS用于分析连接子药物7的纯度。LC-MS分析的条件如下。
管柱:4.6mm×20mm XBridge C18管柱(Waters,Milford USA)
流速:700μL/min
线性梯度:5至70%溶剂B(含有0.1%甲酸的100%乙腈)
溶剂A(ddH2O中0.1%的甲酸)
溶剂B(含有0.1%甲酸的100%乙腈)
如图15所示,在不同储存条件下对连接子药物7进行LC-MS分析的结果显示,当溶解在DMSO中时,连接子药物7是稳定的。
<效价测试>
用ADC处理HER2阴性细胞株MDA-MB-468、具有药性的HER2中等表达乳癌细胞株JIMT-1、和HER2高表达乳癌细胞株BT-474,以分析ADC在这些细胞中的选择性毒性。
将细胞以6×103细胞/孔的密度培养于Corning CellBIND的96孔盘中。于37℃、5%CO2培养箱中培养24小时之后,加入序列稀释的不同浓度的抗体药物复合物(ADC)。接着将细胞放置于37℃、5%CO2培养箱中作用70~120小时。经过70~120小时的培养后,取出旧培养液并洗涤细胞1次后,加入10被稀释的CCK-8试剂于37℃处理4小时。96孔盘放入ELISA读取器侦测在波长450nm的吸收度。通过下式计算细胞存活率:细胞存活率(%)=(样品的密度/对照组的密度)x100%。使用SigmaPlot软体计算ADC样品的IC50值。
根据表3和图16,赫赛汀-连接子药物3、赫赛汀-连接子药物8、和赫赛汀-连接子药物9在HER2阴性细胞株MDA-MB-468中的毒性明显小于商用Kadcyla。
根据表4和图17,赫赛汀-连接子药物14、赫赛汀-连接子药物15、和赫赛汀-连接子药物16在HER2阴性细胞株MDA-MB-468中的毒性明显小于商用Kadcyla。
并且,表3和图18A~18C显示赫赛汀-连接子药物4、赫赛汀-连接子药物7、赫赛汀-连接子药物8、和赫赛汀-连接子药物11对HER2中等表达乳癌细胞株JIMT-1的IC50值小于0.5nM。这些ADC在JIMT-1细胞中的效价比商用Kadcyla好。特别是,图18C显示具有高DAR值(DAR约为8)的赫赛汀-连接子药物11的细胞毒杀性比其他ADC良好。
表4和图19显示赫赛汀-连接子药物14、赫赛汀-连接子药物15、和赫赛汀-连接子药物16对HER2中等表达乳癌细胞株JIMT-1的IC50值小于0.5nM。这些ADC在JIMT-1细胞中的效价比商用Kadcyla好。
此外,表3和图20A~20C显示赫赛汀-连接子药物2、赫赛汀-连接子药物3、赫赛汀-连接子药物4、赫赛汀-连接子药物7、赫赛汀-连接子药物8、赫赛汀-连接子药物9、和赫赛汀-连接子药物11对HER2高表达乳癌细胞株BT-474的IC50值小于0.5nM。这些ADC在BT-474细胞中的效价比商用Kadcyla好。
表4和图21显示赫赛汀-连接子药物14、赫赛汀-连接子药物15、和赫赛汀-连接子药物16对HER2高表达乳癌细胞株BT-474的IC50值小于0.1nM。这些ADC在JIMT-1细胞中的效价比商用Kadcyla好。
这些结果显示表3中具有位点特异性的ADC在HER2中等表达乳癌细胞株JIMT-1和HER2高表达乳癌细胞株BT-474中具有良好的选择性毒性。
表3
表4
<肿瘤生长抑制试验:BT-474异种移植模式>
将1x107的乳癌细胞株BT-474植入NODSCID小鼠皮下,测试ADC于体内(in vivo)的药物效能。
首先,对雌性小鼠植入雌二醇试剂丸(0.36mg/丸;90天释放,Innovative Research of America)。接着将BT-474细胞植入小鼠皮下,并等待肿瘤生长。量测肿瘤的长度和宽度,并以长x宽x宽x1/2(mm3)进行估算及记录肿瘤大小。当肿瘤大小来到359mm3时,于第0天和第21天以静脉注射(10mL/kg B.W.注射体积)空白对照(DPBS)、10mg/kg Kadcyla(购自Roche)、5mg/kg的赫赛汀-连接子药物5、2.5mg/kg的赫赛汀-连接子药物5、5mg/kg的赫赛汀-连接子药物8、2.5mg/kg的赫赛汀-连接子-药物8两次,进行药效实验。
观察小鼠的肿瘤生长和体重,直到第60天。肿瘤生长抑制率(Tumor Growth Inhibition;TGI)计算公式为TGI(%)=[1–(△药物处理组肿瘤体积/△空白对照组肿瘤体积)]x100。期间,若肿瘤大于小鼠体重的10%、肿瘤体积大于1500mm3、或并发其他不良反应,依人道考量主动以二氧化碳将小鼠进行牺牲。
处理ADC经过60天后,经5mg/kg的赫赛汀-连接子药物5、5mg/kg的赫赛汀-连接子药物8、2.5mg/kg的赫赛汀-连接子-药物8处理的组别中,肿瘤生长受到明显的抑制。于第60天,经5mg/kg的赫赛汀-连接子药物5、5mg/kg的赫赛汀-连接子药物8、2.5mg/kg的赫赛汀-连接子-药物8处理的组别的TGI分别为133.8±9.4%、141.7±14%、和142.2±6.7%,而经10mg/kg Kadcyla处理的组别的TGI为约64%(参照表5和图22)。
结果(参照表6)显示给予ADC后各组没有体重下降及其他明显异常临床症状。
表5
表6
<肿瘤生长抑制试验:EC PDX模式>
从台湾大学获得的子宫内膜癌肿瘤组织被证实是HER2阳性(+1)。将肿瘤(直径2~3mm)植入NODSCID小鼠皮下,测试ADC于体内(in vivo)的药物效能。
当肿瘤大小约为100mm3时,小鼠被分为两组,并以静脉注射ADC(10mL/kg B.W.注射体积)。
处理ADC经过12天后,经10mg/kg的赫赛汀-连接子药物7、5mg/kg的赫赛汀-连接子药物7、10mg/kg的赫赛汀-连接子药物11、和5mg/kg的赫赛汀-连接子药物11处理的组别中,肿瘤生长受到明显的抑制。于第12天,经10mg/kg的赫赛汀-连接子药物7、5mg/kg的赫赛汀-连接子药物7、10mg/kg的赫赛汀-连接子药物11、和5mg/kg的赫赛汀-连接子药物11处理的TGI分别为95.6±2.3%、66.9±4.6%、101.7±0.5%、和95.0±3.2%,而经10mg/kg Kadcyla处理的组别的TGI为约26.5±10.1%(参照表7和图23)。
结果(参照表8)显示给予空白对照、10mg/kg Kadcyla、10mg/kg的赫赛汀-连接子药物7、和5mg/kg的赫赛汀-连接子药物7后,没有体重下降及其他明显异常临床症状。虽然在给予10mg/kg的赫赛汀-连接子药物11、和5mg/kg的赫赛汀-连接子药物11后有明显的体重下降,但异常临床症状于一周后开始恢复。
表7
表8
虽然本发明已以数个较佳实施例揭露如上,然其并非用以限定本发明,任何所属技术领域中具有通常知识者,在不脱离本发明的精神和范围内,当可作任意的更动与润饰,因此本发明的保护范围当视后附的权利要求所界定者为准。
- 还没有人留言评论。精彩留言会获得点赞!