用于合成二氧化硅纳米颗粒的方法与流程
本发明涉及用于合成特别适用于诊断和/或治疗的超小二氧化硅纳米颗粒的方法。本发明还涉及获得的超小二氧化硅纳米颗粒。
背景技术:
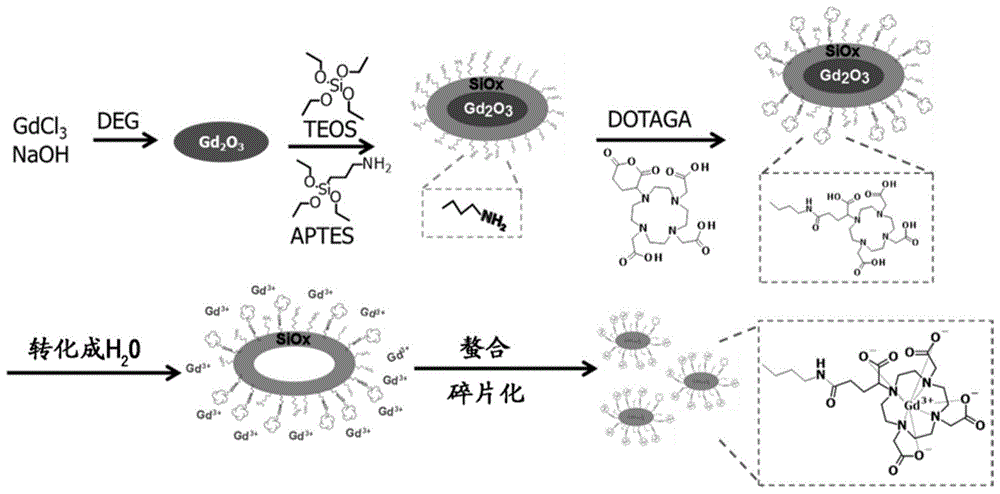
:在世界上许多国家中,癌症是主要的死亡原因。如今,有三种主要的治疗癌症的方法,其是手术、化疗和放疗。放疗意味着使用电离辐射来摧毁肿瘤中的癌细胞。但是,放疗的重大副作用在于辐射还会损害肿瘤周围的正常组织。尽管在波束形状设计和治疗计划方面已经取得了很大的技术进步,但是这种副作用仍然限制了放疗的治疗范围。已经引入了两种主要策略来克服这种副作用:使用不同颗粒(诸如质子、中子或重离子)代替常规的光子,和/或使用放射增敏剂来局部增强肿瘤内的递送剂量。后者为可用作有效增敏剂的新材料的研究打开了大门。对抗癌症的另一个先决条件是精确定位肿瘤。当前,有几种成像技术可用于此目的,包括mri、x射线ct、由单光子发射计算机断层扫描(spect)和正电子发射断层扫描(pet)组成的闪烁扫描法、光学成像和超声成像。这些技术也需要新材料作为探针或造影增强剂,以改进成像质量。此外,每种技术都有其自身的优缺点;因而,将互补技术相结合(即所谓的多模式成像)以实现高的分辨率和灵敏度是一个活跃的研究领域。近年来,纳米技术的出现为开发现代医学(特别是癌症治疗)的新解决方案提供了巨大的机会。这产生了一个迅速发展的领域,称为纳米医学,其中最有前途的研究方向之一是开发纳米材料作为新型治疗剂或用于成像的新型探针或造影剂。这是由于纳米颗粒可以提供众多新优势,诸如:-基于纳米颗粒的化学设计,将大量活性分子保护、递送和释放至某些特定位点的能力;-仅在一个纳米颗粒中结合不同元素(如活性分子、官能团或成像探针)的可能性;-由于epr效应“增强的通透性和滞留性”(这是纳米颗粒在肿瘤中而不是在正常组织中积聚的趋势)而特定地靶向恶性肿瘤的能力;-由其纳米尺寸引起的纳米材料的光学、热、磁和电子性能。在不同类型的纳米颗粒中,包含金属的纳米颗粒引起了很多兴趣。这种类型的纳米颗粒允许利用金属的多种有价值的性能,诸如高z金属(例如au(z=79)、pt(z=78)、hf(z=72)、gd(z=64))的放射增敏能力或钆(gd)的用于mri成像的顺磁性能或核颗粒(例如64cu、68ga、99tc、89zr、111in)放射的用于闪烁扫描成像的性能、90y或177lu的用于居里疗法的性能。文献中已经描述了几个实施例。特别地,已经对二氧化硅纳米颗粒作为用于不同金属的纳米载体进行了深入研究。它们安全且易于合成和官能化。另外,在许多情况下,它们不干扰金属与外部刺激物(例如辐射颗粒、光束等)之间的相互作用。专利申请wo2007124131描述了用螯合剂(即dtta、dtpa)官能化的二氧化硅纳米颗粒,并且在临床中用于螯合gd3+作为造影剂。这些纳米颗粒的流体动力学直径(dh)约为40nm。但是,最近,许多研究表明,建议使用dh小于10nm的超小尺寸,以便通过尿液将纳米颗粒快速、完全地从机体清除。这将防止因导致肾原性系统性纤维化的严重副作用而臭名昭著的金属(如gd3+)的长期毒性。此外,使颗粒尺寸最小化产生更大的表面,因此产生高得多的gd螯合物的负载率。然而,仍然难以合成这种尺寸的纳米颗粒,即具有超小dh的纳米颗粒,例如小于15nm,优选小于10nm。专利申请wo2011135101a2描述了一种基于gd的二氧化硅纳米颗粒,也被称为其显示出用作治疗诊断剂的有前途的结果。该纳米颗粒包含用螯合剂(如dotaga)官能化的聚硅氧烷网络,并且具有小于5nm的dh。颗粒上的大多数螯合剂(通常超过50%)与gd3+形成络合物。该纳米颗粒是使用mri检测肿瘤的非常有效的造影剂。而且,已知其所有组分(即聚有机硅氧烷、dotaga的gd3+络合物)对人是安全的。可通过肾脏清除作用迅速从人体清除,这有助于防止gd3+沉积在器官中,并因此防止称为肾原性系统性纤维化的严重长期并发症。然而,合成这种纳米颗粒并不简单,因为它是经由如图1所示的多步合成完成的。首先,在二甘醇(deg)中合成小的1-3nm的钆氧化物核。然后,通过烷氧基硅烷的水解和缩合反应在该核上涂覆一层聚有机硅氧烷。接下来,用dotaga酸酐对该层进行官能化。之后,将纳米颗粒转移到水中以溶解该核并释放被dotaga螯合的gd3+离子。聚硅氧烷层破碎后,获得纳米颗粒。已经对这种自上而下的方法进行了充分的研究,但是它耗时、耗溶剂且耗原材料。此外,它意味着收率低并且难以改变或添加后合成的不同金属。因而,期望开发可以克服当前合成的限制的更容易的方法来合成该纳米颗粒。特别地,还期望开发一种能够生产超小且稳定的二氧化硅纳米颗粒的一锅合成法。此外,还希望这种新方法能够合成可随后根据应用与不同的金属螯合的空位螯合剂负载率高的纳米颗粒。发明简述本申请的第一方面涉及用于合成二氧化硅纳米颗粒的方法,所述方法包含将至少一种在生理ph下带负电荷的硅烷与以下混合:-至少一种在生理ph下为中性的硅烷,和/或-至少一种在生理ph下带正电荷的硅烷,其中:-中性硅烷与带负电荷的硅烷的摩尔比a定义如下:0≤a≤6,优选0.5≤a≤2;-带正电荷的硅烷与带负电荷的硅烷的摩尔比b定义如下:0≤b≤5,优选0.25≤b≤3;-中性和带正电荷的硅烷与带负电荷的硅烷的摩尔比c定义如下:0<c≤8,优选1≤c≤4。本申请的另一方面涉及平均流体力学直径为0.5-15nm,例如为0.5-10nm二氧化硅纳米颗粒,其包含接枝有螯合剂的聚有机硅氧烷基质,所述螯合剂不含金属离子,并且以至少0.1μmol/mg纳米颗粒的含量存在,优选0.5-2μmol/mg纳米颗粒的含量存在。例如,在一个具体的实施方案中,螯合剂为dotaga,并且螯合剂的含量为0.5-2μmol/mg。发明详述用于合成二氧化硅纳米颗粒的方法第一方面,本申请涉及用于合成二氧化硅纳米颗粒的方法,所述方法包含将至少一种在生理ph下带负电荷的硅烷与以下混合:-至少一种在生理ph下为中性的硅烷,和/或-至少一种在生理ph下带正电荷的硅烷,其中:-中性硅烷与带负电荷的硅烷的摩尔比a定义如下:0≤a≤6,优选0.5≤a≤2;-带正电荷的硅烷与带负电荷的硅烷的摩尔比b定义如下:0≤b≤5,优选0.25≤b≤3;-中性和带正电荷的硅烷与带负电荷的硅烷的摩尔比c定义如下:0<c≤8,优选1≤c≤4。如本文所用,术语“二氧化硅纳米颗粒”是指衍生自硅烷前体的聚合的纳米颗粒。优选地,所述纳米颗粒包含聚有机硅氧烷。所述纳米颗粒还可包含其他化合物,包括有机分子。下文描述了通过该方法获得的二氧化硅纳米颗粒的具体实施方案。如本文所用,术语“生理ph”被认为是7.4。如本文所用,术语“硅烷”是指在硅原子上具有4个取代基的化合物。在优选实施方案中,硅烷选自烷氧基硅烷、羟基硅烷及它们的混合物。可在该方法中使用的硅烷的例子是原硅酸四乙酯(si(oc2h5)4,也称为teos)、原硅酸四甲酯(si(och3)4,也称为tmos)、氨丙基三乙氧基硅烷(h2n(ch2)3-si(oc2h5)3,也称为aptes)、aptes-dotaga、n-(三甲氧基甲硅烷基丙基)乙二胺三乙酸三钠盐((ch3o)3si-(ch2)3n(ch2coona)(ch2)2n(ch2coona)2,也称为taned)和羧基乙基硅烷三醇钠盐((ho)3si-(ch2)2coona,也称为cest)。如本文所用,术语硅烷还包括含有螯合的金属阳离子的任何硅烷化合物。如本文所用,术语硅烷还包括将如下所述的任何官能化剂共价接枝到硅烷前体上的任何硅烷化合物;例如,官能化剂包括荧光团、药物、有机聚合物或靶向配体。如本文所用,术语“烷氧基硅烷”是指式(i)化合物:rnsi(ori)4-n(i)其中:-r为有机基团;-ri为c1-c12烷基,优选为c1-c6烷基;-n为0、1、2或3。根据一个实施方案,n为0或1。如本文所用,术语“羟基硅烷”是指式(ii)化合物:rnsi(oh)4-n(ii)其中:-r为有机基团;-n为0、1、2或3。根据一个实施方案,n为0或1。如本文所用,术语“有机基团”是指经由si-c键与硅原子连接的有机取代基,而与官能化类型无关。有机取代基的实例包括但不限于烷基胺。如本文所用,术语“c1-c12烷基”是指具有1-12个碳原子的直链或支链烷基官能团。合适的烷基包括甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基和叔丁基、戊基及其异构体(例如正戊基、异戊基)以及己基及其异构体(例如正己基、异己基)。根据该方法的一个具体实施方案,纳米颗粒的平均流体动力学直径为0.5-15nm,优选为0.5-10nm。在该方法的具体实施方案中,硅烷(例如选自烷氧基硅烷、羟基硅烷及它们的混合物)可以占试剂总重量的至少80重量%、85重量%或90重量%,该试剂为在反应中用于合成纳米颗粒的起始化合物。该反应可以在质子溶剂(如醇或水溶液)中进行。在一个具体实施方案中,仅用水作为反应溶剂。在其他实施方案中,反应在醇或醇的混合物中进行。可以在该方法中使用的醇包括乙醇、正丙醇、异丙醇、正丁醇、叔丁醇、正戊醇、乙二醇和二甘醇。该反应优选在胶体溶液中进行。这使得能够更好地控制纳米颗粒的直径。典型地,不经由经典溶胶-凝胶工艺进行反应,以避免3个相互连接的凝胶形成。与现有技术的方法相反,本方法的一个优势是它可用一锅合成法进行,而没有中间产物的任何分离或纯化步骤。另一个优点是,特定比率a、b和c的设计使得能够控制二氧化硅纳米颗粒的表面电荷和尺寸,特别是用于生产平均流体动力学直径为0.5-15nm的稳定纳米颗粒。特别地,为了将纳米颗粒的尺寸减小至低于10nm,优选比率a,例如,小于2,并且更优选小于1.5。根据该方法的一个实施方案,所述带负电荷的硅烷包括或基本上由包含至少一个、两个或更多个带负电荷的羧酸官能团的硅烷组成。具有羧酸官能团的带负电荷的硅烷的实例包括但不限于:n-(三甲氧基甲硅烷基丙基)乙二胺三乙酸三钠盐((ch3o)3si-(ch2)3n(ch2coona)(ch2)2n(ch2coona)2,也称为taned)和羧基乙基硅烷三醇钠盐((ho)3si-(ch2)2coona,也称为cest)。根据该方法的另一个实施方案,所述带负电荷的硅烷包括或基本上由包含至少一种螯合剂的硅烷组成。该螯合剂可带有负电荷,例如羧酸官能团。该螯合剂可含有一些螯合的金属阳离子。该螯合剂可选自聚氨基聚羧酸,包括但不限于:-dota(1,4,7,10-四氮杂环十二烷-n,n’,n”,n”’-四乙酸)、dotaga(2-(4,7,10-三(羧甲基)-1,4,7,10-四氮杂环十二烷-1-基)戊二酸)、下式(iii)的do3a-吡啶:-dtpa(二亚乙基三胺五乙酸)、chx-dtpa(反式-环己基-二亚乙基三胺五乙酸)、oxo-do3a(1-氧代-4,7,10-三氮杂环十二烷-4,7,10-三乙酸)、scn-bz-dtpa(对-异硫氰酸根苄基-dtpa)、1b3m(1-(对-异硫氰酸根苄基)-3-甲基-dtpa)、mx-dtpa(1-(2)-甲基-4-异氰酸根苄基-dtpa);-edta(2,2',2”,2”'-(乙烷-1,2-二基二氮基)四乙酸);-egta(乙二醇-双(2-氨基乙基醚)-n,n,n′,n′-四乙酸)、bapta(1,2-双(邻氨基苯氧基)乙烷-n,n,n′,n′-四乙酸);-nota(1,4,7-三氮杂环壬烷-1,4,7-三乙酸);-pcta(3,6,9,15-四氮杂双环[9.3.1.]十五碳-1(15),11,13-三烯-3,6,9-三乙酸);-下式(iv)的tmpac;-及它们的混合物。根据该方法的一个实施方案,该混合步骤还可包括在生理ph下为中性的硅烷,所述中性硅烷可包含螯合剂。例如,包含在中性硅烷中的螯合剂可选自卟啉、氯、1,10-菲咯啉、联吡啶和三联吡啶。根据该方法的一个实施方案,通过该方法获得的纳米颗粒包含大于0.1μmol/mg的螯合剂。例如,通过该方法获得的纳米颗粒可包含大于0.5μmol/mg的螯合剂,或0.1-5μmol/mg的螯合剂,或0.5-2μmol/mg的螯合剂。根据该方法的一个具体实施方案,该方法包括使用具有螯合剂的硅烷,该螯合剂不含金属离子。例如,试剂的总螯合剂的小于5mol%、1mol%或0.1mol%与金属离子形成络合物。在这种情况下,取决于所需的应用,可以在第二步骤中通过感兴趣的金属离子将通过该方法获得的具有游离螯合剂的纳米颗粒螯合。或者,该方法包括使用具有螯合剂的硅烷,该螯合剂螯合金属离子。例如,试剂的总螯合剂的大于50mol%、70mol%或90mol%与金属离子形成络合物。在这种情况下,通过该方法获得的纳米颗粒具有螯合的金属离子,并且可在不进一步添加金属离子的情况下直接使用。可被螯合剂螯合的金属离子包括碱金属离子及它们的放射性同位素、过渡金属离子及它们的放射性同位素、后过渡金属离子及它们的放射性同位素、稀土金属离子及它们的放射性同位素、及它们的混合物。过渡金属包括hf、cu、pt、au、tc、y、mn、ru、fe、zr及它们的混合物。后过渡金属包括bi、ga、in及它们的混合物。稀土金属包括镧系元素,诸如gd、dy、eu、tb、nd、yb、er、ho、lu或它们的混合物,更优选为gd。例如,放射性同位素将选自ac、th、pa、np、u和pu。例如,gd、dy、mn和fe适用于生产在mri中用作造影剂的纳米颗粒。例如,eu、tb、nd、yb和er适用于用作荧光剂的纳米颗粒。例如,ho、bi、y和lu适用于用作居里疗法治疗剂的纳米颗粒。例如,lu、yb、gd、bi、hf和ho适用于用作放射增敏剂的纳米颗粒。例如,cu、ga、tc、y、in和zr适用于用作闪烁扫描探针的纳米颗粒。根据该方法的具体实施方案,与纳米颗粒的总重量相比,通过该方法获得的纳米颗粒包含大于10重量%,例如大于15重量%,或10重量%-20重量%的金属离子。该含量可通过对冻干粉末的元素分析来测定。根据该方法的具体实施方案,通过该方法获得的纳米颗粒包含大于0.1μmol/mg,例如大于0.25μmol/mg或0.1-1.5μmol/mg的金属离子。根据一个实施方案,该混合步骤包含至少一种带正电荷的硅烷,所述带正电荷的硅烷包含至少一种带正电荷的氨基官能团。具有氨基官能团的带正电荷的硅烷的实例包括但不限于:aptes。在该方法的具体实施方案中,合成纳米颗粒导致通过形成硅氧烷桥si-o-si而产生聚有机硅氧烷网络。这些硅氧烷桥通过羟基硅烷的缩合和脱水而获得。如果在本申请的方法中使用烷氧基硅烷,则该反应在水溶液中进行以首先将烷氧基硅烷水解为羟基硅烷。在这种情况下,纳米颗粒具有聚有机硅氧烷基质。根据一个具体实施方案,根据本申请的方法包含将至少一种在生理ph下带负电荷并且包含至少一种选自聚氨基聚羧酸的螯合剂的羟基硅烷或烷氧基硅烷与以下混合:-至少一种在生理ph下为中性的羟基硅烷或烷氧基硅烷,和/或-至少一种在生理ph下带正电荷并且包含氨基官能团的羟基硅烷或烷氧基硅烷,其中:-中性硅烷与带负电荷的硅烷的摩尔比a定义如下:0≤a≤6,优选0.5≤a≤2;-带正电荷的硅烷与带负电荷的硅烷的摩尔比b定义如下:0≤b≤5,优选0.25≤b≤3;-中性和带正电荷的硅烷与带负电荷的硅烷的摩尔比c定义如下:0<c≤8,优选1≤c≤4。根据一个具体实施方案,根据本申请的方法包含将至少一种在生理ph下带负电荷的烷氧基硅烷(所述烷氧基硅烷选自aptes-dotaga、taned、cest及它们的混合物)与以下混合:-至少一种在生理ph下为中性的烷氧基硅烷,所述烷氧基硅烷选自tmos、teos及它们的混合物;和/或-在生理ph下带正电荷的aptes,其中:-中性硅烷与带负电荷的硅烷的摩尔比a定义如下:0≤a≤6,优选0.5≤a≤2;-带正电荷的硅烷与带负电荷的硅烷的摩尔比b定义如下:0≤b≤5,优选0.25≤b≤3;-中性和带正电荷的硅烷与带负电荷的硅烷的摩尔比c定义如下:0<c≤8,优选1≤c≤4。根据一个具体实施方案,根据本发明的方法包含将在生理ph下带负电荷的aptes-dotaga与以下混合:-至少一种在生理ph下为中性的烷氧基硅烷,所述烷氧基硅烷选自tmos、teos及它们的混合物,和/或-在生理ph下带正电荷的aptes,其中:-中性硅烷与带负电荷的硅烷的摩尔比a定义如下:0≤a≤6,优选0.5≤a≤2;-带正电荷的硅烷与带负电荷的硅烷的摩尔比b定义如下:0≤b≤5,优选0.25≤b≤3;-中性和带正电荷的硅烷与带负电荷的硅烷的摩尔比c定义如下:0<c≤8,优选1≤c≤4。官能化剂通过该方法获得的纳米颗粒可以有利地用特定的有机分子官能化,例如用于治疗或成像中的具体医学应用。因此,在该方法的具体实施方案中,可用包含至少一种官能化剂的至少一种硅烷进行该混合步骤。如本文所用,术语“官能化剂”是指通过共价缀合连接到硅烷上的有机分子,该有机分子用于将获得的纳米颗粒进行特定官能化。这种官能化剂包括但不限于荧光团、药物、有机聚合物或靶向配体。因此,在该方法的具体实施方案中,可用包含至少一种荧光团的至少一种硅烷进行该混合步骤。优选地,包含荧光团的硅烷与中性硅烷的摩尔比d定义如下:0.001≤d≤0.2。可通过以下方法获得这种包含荧光团的硅烷:将具有反应性部分的荧光化合物与二氧化硅前体共价缀合以获得荧光二氧化硅前体,然后使荧光二氧化硅前体与硅烷(诸如四烷氧基硅烷)反应,从上部形成包含至少一种荧光团的硅烷。可使用的荧光化合物包括但不限于cy5、cy5.5、cy7、异硫氰酸荧光素(fitc)、四甲基罗丹明异硫氰酸酯、x-罗丹明、alexa、bodipy荧光染料、cw800和吲哚菁绿(icg)。根据另一个实施方案,该混合步骤包含至少一种包含至少一种药物部分的硅烷。优选地,包含药物的硅烷与中性硅烷的摩尔比e定义如下:0.1≤e≤5。根据一个具体实施方案,与纳米颗粒的总重量相比,该纳米颗粒包含0.5-50重量%,例如2-10重量%的药物部分。这种包含药物部分的硅烷还可通过以下方法获得:例如如抗体-药物-缀合物的文献中所述的,首先制备接头-药物部分,然后通过共价缀合使接头-药物部分与至少一种硅烷反应以形成所述包含至少一种药物部分的硅烷。接头的实例包括可裂解的接头,诸如对氨基苄氧基羰基(pabc)基团。可用于制备包含药物部分的硅烷的药物的实例包括但不限于小分子药物,例如化学治疗药物,诸如烷化剂、蒽环类、紫杉烷类、hdac抑制剂、拓扑异构酶i或ii的抑制剂、激酶抑制剂、核苷酸类似物和前体类似物。可用于制备包含药物部分的硅烷的药物的具体实例包括但不限于放线菌素、全反式视黄酸(aciderétinoiqueall-trans)、阿扎胞苷、硫唑嘌呤、博来霉素、硼替佐米、卡铂、卡培他滨、顺铂、苯丁酸氮芥、环磷酰胺、阿糖胞苷、柔红霉素、多西他赛、多西氟尿啶、多柔比星、表柔比星、埃博霉素、依托泊苷、氟尿嘧啶、吉西他滨、羟基脲、伊达比星(darubicine)、伊马替尼、伊立替康、甲氯乙胺(mechlorethamine)、巯嘌呤、甲氨蝶呤、米托蒽醌、奥沙利铂、紫杉醇、培美曲塞、替尼泊苷、硫鸟嘌呤(tioguanine)、拓扑替康、戊柔比星(valrubicin)、威罗非尼、长春碱、长春新碱、长春地辛、来那度胺、依鲁替尼、阿比特龙、厄洛替尼、依维莫司、尼洛替尼、舒尼替尼、索拉非尼、戈舍瑞林、那达铂(nedaplatine)、laboplatine、heptaplatine、及它们的混合物。根据另一个实施方案,该混合步骤包括至少一种包含至少一种靶向配体的硅烷。如本文所用,靶向配体是与硅烷连接的分子,其有助于在体内将纳米颗粒靶向特定的细胞组分。这种配体可在体内靶向特定的器官、组织或细胞类型,例如用于特定的医学或诊断应用。例如,这种靶向配体可以是肽、蛋白质、糖(例如凝集素)、生物聚合物、合成聚合物、抗原、抗体、适体和纳米抗体。靶向配体的实例包括但不限于赫赛汀(曲妥珠单抗)、美罗华(利妥昔单抗)、cd19抗体、哌他尼布(pepaptanib)、a10适体、crgd肽、atwlppr肽、vap、lyp-1、转铁蛋白、lfrh、叶酸、半乳糖或asgpr靶向配体、生物素、甘露糖。根据另一个实施方案,该混合步骤包括至少一种包含至少一种有机聚合物的硅烷。例如,该有机聚合物可以是peg(聚乙二醇)、聚乳酸(polylactate)、聚乳酸(polylacticacids)、糖(sugards)、脂质、聚谷氨酸(pga)、聚乙醇酸、聚(乳酸-乙醇酸)共聚物(plga)、聚乙酸乙烯酯(pva)及它们的组合。已经表明,与现有技术方法相反,本方法能够生产超小且稳定的纳米颗粒,而无需使用稳定的有机聚合物。因此,在一个具体实施方案中,本申请的方法不包括接枝到硅烷的任何有机聚合物。二氧化硅纳米颗粒本发明还涉及通过上述方法获得的二氧化硅纳米颗粒。典型地,通过该方法获得的二氧化硅纳米颗粒的平均流体动力学直径有利地为0.5-15nm,例如1-10nm,或1-5nm,或2-7nm。根据本申请的教导,本领域技术人员将能够调节该方法中定义的特定比率a、b和c以获得二氧化硅纳米颗粒的所需尺寸。如本文所用,术语“平均流体力学直径”旨在表示颗粒的流体力学直径的谐波平均值。测量该参数的方法是通过光子相关光谱法,该方法也描述在标准iso13321:1996中。通过该方法获得的二氧化硅纳米颗粒还可包含荧光化合物、螯合剂(具有或不具有金属)、药物部分、靶向配体或共价连接的有机聚合物。在具体实施方案中,通过该方法获得的这种二氧化硅纳米颗粒具有eu、cu、gd、tb、ho、bi或它们的混合物作为被螯合剂螯合的金属离子。在具体实施方案中,通过该方法获得的这种二氧化硅纳米颗粒具有gd作为被螯合剂螯合的金属离子。在具体实施方案中,通过该方法获得的这种二氧化硅纳米颗粒具有bi、hf或它们的混合物作为被螯合剂螯合的金属离子。在具体实施方案中,通过该方法获得的这种二氧化硅纳米颗粒具有lu、y、cu、zr、in、ga或它们的混合物作为被螯合剂螯合的金属离子。在具体实施方案中,通过该方法获得的这种二氧化硅纳米颗粒具有稀土金属离子作为被螯合剂螯合的金属离子。在具体实施方案中,通过该方法获得的纳米颗粒不包含结晶核,例如可在具有核-壳结构的纳米颗粒中发现的。例如,通过本方法获得的纳米颗粒不包含金属、氧化物、硫化物、氟化物或碳化物的结晶核。在本发明的一个实施方案中,纳米颗粒不包含金属核,诸如gd氧化物核,无论其是否结晶。根据一个实施方案,二氧化硅纳米颗粒不包含聚乙二醇(peg)壳。本申请还涉及平均流体力学直径为0.5-15nm的二氧化硅纳米颗粒,其包含接枝有螯合剂的聚有机硅氧烷基质,所述螯合剂不含金属离子,并且以至少0.1μmol/mg纳米颗粒,优选0.5-2μmol/mg纳米颗粒的含量存在。例如,在一个具体实施方案中,螯合剂是dotaga并且螯合剂的含量为0.5-2。不含金属离子的螯合剂的含量可以通过hplc测定,或例如通过直接用铕或钆与二甲苯酚橙组合滴定来测定。二氧化硅纳米颗粒的用途根据本申请的纳米颗粒可以优选用于治疗或诊断方法中或用作治疗诊断剂。例如,该纳米颗粒可用作治疗剂,诸如用于放疗或中子疗法的增敏剂或放射源、用于光动力疗法(pdt)的试剂或用于治疗分子(诸如化学治疗剂)的递送剂。纳米颗粒还可有利地用作多模态成像剂,其可在磁共振成像(mri)中或在具有单光子发射计算机断层扫描(spect)或正电子发射断层扫描(pet)的闪烁扫描中或在荧光光学成像中、或在x射线计算机断层扫描(x射线ct)中或结合了这些技术中的至少两种的多模态成像中用作造影剂。具有游离螯合剂的纳米颗粒还可用作体内有毒金属的螯合剂,例如hg、pb、al、cd或cr的螯合剂。具有游离螯合剂的纳米颗粒还可用于调节金属稳态,特别是调节内源金属,如fe、cu、zn或mn,或调节外源金属,如hg、pb、al、cd或cr。因此,本申请涉及药物组合物,其包含通过上述定义的方法获得的治疗有效量的二氧化硅纳米颗粒与药学上可接受的载体的组合。本申请还涉及在人或动物中成像的方法,包含以下步骤:(i)施用通过本申请获得的纳米颗粒,作为t1mri造影剂,(ii)使用适当的mri序列捕获图像。本申请还涉及通过放疗治疗需要其的患者的方法,其包含以下步骤:(i)施用通过本申请获得的纳米颗粒,作为放疗的增敏剂,(ii)照射患者以进行放疗。对于该特定应用,本申请的纳米颗粒优选包括gd,其作为与螯合剂螯合的放射增敏剂。附图简要说明图1示出了如wo2011135101a2中报道的合成纳米颗粒的反应图式。图2示出了根据实施例1的纳米颗粒的dls图(图2a)和zeta电位(图2b)。图3示出了cest、taned和根据实施例1的纳米颗粒在d2o中的1h-nmr谱。图4示出了根据实施例1的纳米颗粒在d2o中的2d-nmr(dosy)光谱。图5示出了根据实施例1的纳米颗粒的固态29si-nmr光谱。图6示出了根据实施例2的步骤1.a合成aptes-dotaga的反应图式。图7示出了根据实施例2的步骤1.b合成aptes-dotaga的反应图式。图8示出了40μmeucl3和100μmdotaga混合物在395nm处激发的发射光谱(图8a)以及根据实施例2的步骤1.b的aptes-dotaga合成混合物在594nm处(圆形)和616nm处(方形)处的滴定曲线(图8b)。图8c示出了根据实施例2的步骤1b的dotaga的ir光谱。图8d示出了根据实施例2的步骤1b的aptes-dotaga的反应混合物的ir光谱。图9示出了自下而上合成纳米颗粒的反应图式:根据实施例2的步骤2的策略a(实心箭头)和策略b(虚线箭头)。图10示出了在与gd3+螯合之前,根据实施例2的步骤2.a的uchsnp-1纳米颗粒的dls图。图11示出了根据实施例2的步骤2.a的uchsnp样品在395nm处激发的在594nm处(圆形)和616nm处(方形)的滴定曲线。图12示出了uchsnp-1的nmr光谱:nmr-dosy光谱(图12a);127g/luchsnp-1的1hnmr光谱(图12b);nmr-dosy光谱(图12c);127g/luchsnp-1@lu的1hnmr谱图(图12d),以及根据实施例2的步骤2.a的颗粒上的aptes和aptes-dotaga官能团上h1、h2和h3的位置(图12e)。图13示出了与gd3+螯合后从冻干粉末中再分散的根据实施例2的步骤2.a的uchsnp@gd-1的dls图和zeta电位。图14示出了浓缩之前的根据实施例2的步骤2.b的uchsnp@gd-2纳米颗粒的dls图。图15示出了纯化后从冻干粉末中再分散的根据实施例2的步骤2.b的uchsnp@gd-2纳米颗粒的dls图和zeta电位。图16示出了根据实施例2的步骤2.b的uchsnp@gd-2样品在395nm处激发的在594nm处(圆形)和616nm处(方形)的滴定曲线。图17示出了根据实施例3的一锅合成法合成纳米颗粒并通过调节硅烷的比率来控制尺寸的反应图式。图18示出了根据实施例3的aptes与dotaga酸酐反应后的混合物(图32a)和在ph9下暴露过夜的相同混合物(图32b)的eu滴定曲线。图19示出了根据实施例3的具有不同比率起始硅烷的纳米颗粒的dls图。图20示出了根据实施例3的uchsnp-3(图20a)、uchsnp-4(图20b)、uchsnp-5(图20c)和uchsnp-6(图20d)的eu滴定曲线。图21示出了根据实施例4的在合成期间不同步骤下uchsnp-7的dls图:在deg中aptes+dotaga酸酐(虚线,方形);在deg中aptes+dotaga酸酐+teos(虚线,上三角);在水中aptes+dotaga+teos(虚线,下三角);在水中aptes+dotaga+teos,其通过0.2μm膜过滤(直线,圆形)(图21a);纯化后(图21b)。图22示出了根据实施例4的uchsnp-7纳米颗粒的eu滴定曲线。图23示出了根据实施例4的uchsnp-7@m的dls图(m:gd(方形)、tb(圆形)、ho(上三角)和bi(下三角))。图24a示出了根据实施例4的uchsnp-7在不同ph下的zeta电位的完整曲线。图24b示出了根据实施例4的uchsnp-7@gd、uchsnp-7@tb、uchsnp-7@ho和uchsnp-7@bi在ph6.6下的zeta电位。图25a示出了根据实施例4的uchsnp-7、uchsnp-7@gd、uchsnp-7@tb、uchsnp-7@ho和uchsnp-7@bi的紫外可见吸收光谱。图25b示出了根据实施例4的uchsnp-7、uchsnp-7@ho和在0.1mmhcl中的50mmhocl3的紫外可见吸收光谱。图25c示出了根据实施例4的uchsnp-7@gd的紫外可见激发和发射光谱。图25d示出了根据实施例4的uchsnp-7@tb的紫外可见激发和发射光谱。图26示出了根据实施例4的uchsnp-7、uchsnp-7@gd、uchsnp-7@tb、uchsnp-7@ho和uchsnp-7@bi的红外光谱。图27a示出了注射根据实施例5的uchsnp@gd-2前(左)和后(右,直至6h)的肿瘤组织(白色箭头)的mri截面。图27b示出了注射根据实施例5的uchsnp@gd-2后,肿瘤组织中的动态mri信号增强。图27c示出了注射根据实施例5的uchsnp@gd-2后,肝中的动态mri信号增强。实施例材料和方法材料盐酸(hcl,37%)购自vwrchemicalsbdhprolabo(法国)。氢氧化钠小球(naoh,≥98%)购自sigma-aldrichchemicals(法国)。制备不同浓度2m-10-4m的盐酸和氢氧化钠水溶液,以调节溶液的ph值。六水合氯化铕(eucl3.6h2o,99.9%)、六水合氯化镥(lucl3.6h2o,99.9%)、六水合氯化铽(tbcl3.6h2o,99.9%)、六水合氯化钬(hocl3·6h2o,99.9%)、原硅酸四乙酯(si(oc2h5)4,teos,98%)、氨丙基三乙氧基硅烷(h2n(ch2)3-si(oc2h5)3,aptes,99%)、用于合成硅烷前体的无水dmso、用于nmr实验的氧化氘d2o、用于制备ph为5的缓冲液的冰醋酸、blackt(ebt)和用于络合滴定的ph10的氨水缓冲液购自sigma-aldrichchemicals(法国)。n-(三甲氧基甲硅烷基丙基)乙二胺三乙酸三钠盐((ch3o)3si-(ch2)3n(ch2coona)(ch2)2n(ch2coona)2,taned,水中含量为45%)和羧基乙基硅烷三醇钠盐((ho)3si-(ch2)2coona,cest,水中含量为25%)购自abcrgmbh(德国)。1,4,7,10-四氮杂环十二烷-1-戊二酸酐-4,7,10-三乙酸(dotaga酸酐)由chematech(法国)提供。六水合氯化钆(gdcl3.6h2o,99.999%)购自metallrareearthlimited(中国)。milli-q水(ρ>18mω)用作水源。浓缩器和200盒(mwco=3kda或5kda)购自sartoriusstedimbiotech(法国)。方法动态光散射(dls)和zeta电位用来自malverninstruments的zetasizernano-s(633nmhe-ne激光)通过dls测量纳米颗粒的流体动力学直径分布。用一次性pmma比色杯(carlrothgmbh,德国)对0.5-1ml溶液进行测量。通过该设备优化了衰减器和位移。为了测定zeta电位,在每次测量前,将冻干粉末再分散在水中以获得100mg/ml的溶液,且将其在含有5mmnacl的水溶液中稀释至10-18mg/ml并调节至所需的ph。在20℃下、dts1061折叠毛细管池(malverninstrumentsltd,美国)中记录zeta电位测量。基于smoluchowski方程由电泳迁移率自动计算得出zeta电位(ζ),ν=(εε0ζ/η)ζ,其中ν是测得的电泳迁移率,η是粘度,ε是电解液的介电常数,ε0≈8.854x10-12c2n-1m-2是真空介电常数。在每次测量前,将冻干粉末再分散在水中以获得100mg/ml的溶液,且将其在含有5mmnacl的水溶液中稀释至10mg/ml并调节至所需的ph。色谱法方法1:测定纳米颗粒的纯度并定量纳米颗粒上的游离dotaga使用具有cbm-20a控制器总线模块的shimadzuprominence系列uflc系统、lc-20ad泵、cto-20a柱温箱和spd-20a紫外可见检测器进行梯度hplc分析。检测波长设定为295nm,其中只有有机螯合剂可以高度吸收以表征空位纳米颗粒,或者设定为700nm,其中dotaga的铜络合物特异性吸收以表征纳入铜的纳米颗粒。当表征纳入钆的纳米颗粒时,添加fr-20a荧光检测器((λex=274nm,λem=312nm)以检测来自gd络合物的荧光信号。将柱温保持在30℃。用两个流动相进行梯度lc洗脱:(a)milli-q水/tfa99.9:0.1v/v和(b)乙腈(ch3cn)/tfa99.9:0.1v/v。每次将20μl的样品上样至注射阀,并以1ml/min的流速注入jupiterc4色谱柱(150mm×4.60mm,5μm,phenomenex)。然后,将洗脱程序编程如下:在7min内用1%的溶剂b洗脱反应物和片段,然后在15min内用1%-90%的梯度洗脱纳米颗粒。在7min内维持b的浓度。然后,在1min内将溶剂b的浓度降至1%,并保持8min来重新平衡该系统,以进行新的分析。在测量每个样品之前,在相同条件下通过注入milli-q水获得基线。通过将颗粒峰下的面积除以颗粒峰和反应物下的总面积来计算纯度。该方法还用于以cu2+为探针定量纳米颗粒上游离dotaga的含量。将过量的cuso4添加到已经将ph值调节为小于或等于3的超小杂化螯合二氧化硅纳米颗粒(uchsnp)的溶液中。络合可能会降低溶液的ph值。因而,在80℃下温育至少2h之前,应将ph值重新调节至稳定在3。使用在700nm的可见检测器以特异性检测游离或接枝在纳米颗粒上的铜络合物的吸收。通过将峰面积与其在不同浓度(cu2+4mm-32mm,dotaga(cu2+)0.1mm-15mm)下的校准曲线进行比较来测定cu2+和dotaga(cu2+)的浓度。游离cu2+和dotaga(cu2+)的总浓度可以求和,以验证引入的量。游离dotaga的含量(mol/g)可以通过其摩尔浓度(mol/l)和分析样品的质量浓度(mg/l)来计算。方法2:由dotaga酸酐合成后,鉴定aptes-dotaga使用与方法1中所述的相同系统进行等度hplc分析。在这种情况下,荧光检测器(λex=274nm,λem=312nm)是检测gd络合物信号的主要检测器。流动相固定在100%(a)和0%(b)上,以减慢gd3+、aptes-dotaga(gd3+)和dotaga(gd3+)的洗脱并清楚地分离它们。以与方法1相同的方式引入样品。保持流动10min以洗脱所有预期的峰。之后,将溶剂b逐渐升至100%,以洗涤样品中或柱的溶剂中积聚的杂质。然后,在1min内将溶剂b的浓度降至0%,并在15min内保持该浓度,来重新平衡该系统,以进行新的分析。在测量每个样品之前,以与前一种方法相同的方式获得基线。将合成产物溶解在水中,并与gdcl3混合,以在ph约为6下使合成混合物和gd3+的最终浓度分别达到5g/l和10mm,并在37℃下温育该合成产物15h以使其络合。分析后,收集洗脱液用于质谱分析(ms)。将洗脱溶液冻干以去除溶剂和过量的tfa。将冻干粉末以比冻干之前高一倍的浓度再分散在水中,以确保样品足以进行ms分析。在相同条件下,用hplc分析ph为6的10mmgdcl3样品和ph为5的2mmdotaga(gd3+)加1mmgdcl3的混合物,以通过滞留时间(tr)鉴定gd3+和dotaga(gd3+)的峰。方法3:定量由dotaga酸酐合成后的硅烷前体aptes-dotaga的量通过与方法2中相同的系统和荧光检测器设定进行等度hplc分析。以相同的方式引入样品。但是,使用bds-hypersil-c18色谱柱(250mm×4.60mm,5μm,thermofisherscientific)代替c4色谱柱,以增加小分子的分离能力。此外,流动相固定为99%(a)和1%(b)而不是100%(a),以避免峰的tr轻度波动。该波动归因于“疏水塌陷”,因为溶剂不足够疏水以润湿静态相的表面。保持流动25min,以洗脱所有预期的峰。之后,出于与上述相同的目的,将溶剂b逐渐升高至100%。然后,在进行新分析之前重新平衡该系统。在测量每个样品之前,以相同的方式获得基线。将合成产物溶于ph为5的乙酸盐缓冲液中。向该溶液中添加ph4的50mmgdcl3溶液,以使合成混合物和gd3+的最终浓度分别为57.8mg/l和0.2mm。将该溶液在80℃下温育48h,以完成络合。最终溶液是透明的,但通过0.2μm膜进行过滤,以确保灰尘的大颗粒不会阻塞hplc色谱柱。在相同条件下分析了ph4的1mmgdcl3样品和ph5.7的0.05mmdotaga(gd3+)混合物,以通过tr鉴定gd3+和dotaga(gd3+)的峰。通过将其峰面积与不同浓度(0.01mm-0.15mm)下的校准曲线进行比较,来测定dotaga(gd3+)的浓度。通过将使用eu磷光通过滴定测定的aptes-dotaga和dotaga的总浓度减去dotaga(gd3+)的浓度,间接测定aptes-dotaga(gd3+)的浓度。未反应的dotaga和aptes-dotaga的含量(mol/g)可以从它们的摩尔浓度(mol/l)和所分析的合成混合物的质量浓度(mg/l)来计算。质谱(ms)ms用于鉴定hplc色谱图中aptes-dotaga(gd3+)和dotaga(gd3+)的峰。质谱以负模式记录在飞行时间质谱仪microtof-qii(brukerdaltonics,德国)上。1h核磁共振(nmr)和扩散有序光谱(dosy)所有实验均在配备5mmbbfo和bbi探针的brukeravanceiii500mhz光谱仪上、298k且不旋转下进行。将冻干的二氧化硅纳米颗粒分散在d2o中。对于1hnmr扩散实验,使用标准ledbpgp2s序列。扩散延迟d20设定为100ms,且双极性脉冲p30调节为在全强度下获得95%的衰减(典型地在2-4ms的范围内)。在扩散维度上获得了32或64个点。比较使用标准dosy2d指令、dynamiccenter和nmrnotebook程序获得的处理数据,使用nmrnotebook获得了最佳结果,即使在同一化学位移下混合了多个信号,其也提供了良好的数据拟合。所报道的流体动力学直径(dh)可用众所周知的stokes-einstein公式简单地从扩散系数(d)得出:dh=kbt/3πηd,其中kb是玻尔兹曼常数,t是绝对温度,η是溶剂粘度(对于298k下的d2o为1.13cp)。固态29sinmr光谱固态29sinmr实验是在带有mas4mm双h/x探针的brukeravance500wb光谱仪上进行的,mas速率为10khz,光谱频率为99.34mhz。用高功率去耦mas脉冲序列获得定量谱,其脉冲长度为4μs(对应于90°脉冲),在1200次采集扫描中的重复延迟为240s。用dmfit软件进行光谱分解。可将信号反卷积为对应于六个不同si环境的六个贡献。它们有两种主要类型:csi(osi)no3-n和si(osi)mo4-m,通常分别标记为tn(对于第三级)和qm(对于第四级)。tn类是由有机三烷氧基硅烷(诸如cest、aptes、taned或aptes-dotaga)形成的,而qm是由四烷氧基硅烷(诸如teos)形成的。eu磷光滴定通过eu磷光滴定是精确定量aptes-dotaga合成混合物中和冻干最终粉末中螯合剂含量(mol/g)的主要方法。将合成混合物或冻干粉末再分散在水中。在ph5的醋酸盐缓冲液中制备一定量的该溶液和eucl3含量增加的一系列样品。在测量之前,将这一系列样品在80℃下温育48h。使用variancaryeclipse荧光分光光度计在分辨时间模式下进行磷光测量。对于单次读取测量,参数设定如下:激发波长为395nm;发射波长为594nm和616nm,这是eu3+离子的特征激发和发射;激发狭缝为10nm;发射狭缝为10nm;延迟时间0.2ms;总衰减时间0.02s;平均时间5s;门控时间5ms;闪光次数1;激发滤光片335–620nm;发射滤光片550-1100nm;高电压。为了扫描发射光谱,使用分辨率为1nm的类似参数,除了将平均时间减少到1s以加快测量速度。当发光强度不再随eu3+的添加量线性增加时测定终点。弛豫度测量弛豫度测量在minispecmq60nmr分析仪(brucker,美国)上37℃、1.4t(60mhz)下进行。在特定的gd3+浓度(mm)下测量样品,该浓度由icp-oes或元素分析测量。测量了纵向弛豫时间t1和横向弛豫时间t2(s)。然后,根据下式获得弛豫度ri(s-1.mm-1)(i=1,2):i=1或2。元素分析元素分析用filabsas.,dijon,法国进行,其能够测定粉末样品中gd、c、n和si的含量。电感耦合等离子体-光发射光谱法(icp-oes)通过电感耦合等离子体-光发射光谱仪(icp-oes)(varian710-es光谱仪,美国)测定纳米颗粒中金属的准确浓度。将dls测量后的溶液重新用于该测量。将金属(gd、tb、ho或bi)的估计浓度为10ppm的颗粒的溶液在80℃的4-5ml王水(hno367%与hcl37%(1/2;v/v))中消解3h。随后,将混合物用精确的50mlhno35%(v/v)稀释至100、200和400ppb。在分析之前,使这些溶液通过0.2μm膜过滤。通过用hno35%(w/w)连续稀释,由1000ppmgd、tb、ho和bi标准溶液制备校准样品。对于gd样品,选定测量波长为342.246、335.048、336.224nm;对于tb样品,选定测量波长为350.914、367.636、387.417nm;对于ho样品,选定测量波长为345.600、339.895、341.644nm;对于bi样品,选定测量波长为223.061nm。结果是三个样品在不同选定波长下的平均值大概为100、200和400ppb。紫外可见光谱用variancary50分光光度计(美国)记录紫外可见光谱。在5g/l下测量uchsnp-7和uchsnp-7@ho的溶液,在0.06g/l下测量uchsnp-7、uchsnp-7@m(m:gd、tb、ho、bi)的溶液。红外光谱(ir)用iraffnity-1shimadzu进行红外光谱分析。透射模式与happ-genzel切趾函数结合使用,30次扫描、分辨率为4cm-1,范围为400-4000cm-1。在冻干之前,将溶液的ph调节至2。记录所获粉末的光谱。实施例1:使用taned、cest和teos在水中合成超小二氧化硅纳米颗粒将taned(8.22ml,8mmol)和cest(5.57ml,8mmol)添加到水(63ml)中,并在室温下搅拌15min。然后,将teos(5.57ml,16mmol)添加到上述溶液中。将其在室温下搅拌过夜,以使溶液变得均匀。之后,通过加入几滴适当浓度的hcl将溶液的ph从10.5降低至7.4。将溶液搅拌24h,然后将其从ph7.4重新调节至ph4.5。将溶液搅拌6h,然后置于烘箱,在80℃下静置一晚。少量溶液通过0.2μm膜过滤,并通过动态光散射(dls)和高效液相色谱(hplc)进行分析。然后,以10-4mhcl溶液为溶剂,将整个溶液通过vivaspintm(mwco=3kda)过滤,进行纯化。将该溶液引入20mlvivaspin管中,并离心直至剩余一半体积(纯化速率21=2)。通过用10-3m盐酸溶液填充试管并再次离心,重复此步骤几次,直到从hplc色谱图计算出的纯度达到≥90%(通常,28=256的纯化速率)为止。然后,使该溶液通过0.2μm膜过滤以去除最大的杂质。最后,将溶液冻干以长期保存。用流体力学尺寸、zeta电位和组成来表征获得的颗粒。图2-a示出了通过dls测量的最终颗粒的尺寸分布。平均尺寸约为4.6nm,标准偏差为1.1nm。图2-b示出了zeta电位作为通过在同一仪器中集成的激光多普勒测速仪测得的ph值的函数。由于聚硅氧烷在碱性ph下的稳定性降低,因此在ph8下停止测量。最终颗粒具有预期的负表面电荷。ph7时的zeta电位为-25.8mv。根据方法1进行hplc分析。基于在295nm处的吸收,最终纳米颗粒的纯度为92.4%。图3示出了d2o中cest、taned和最终纳米颗粒的1h核磁共振(nmr)光谱。该结果表明cest和taned被纳入最终纳米颗粒中。图4示出了通过在d2o中的最终纳米颗粒样品上应用扩散有序光谱法(dosy)技术创建的2dnmr光谱。该技术使我们能够测量与每个1h信号相关的扩散系数(dh)。在图4中,x轴显示化学位移(ppm),在该化学位移中找到了先前在1d1h光谱中观察到的所有1h峰。y轴显示dh的值。然后,根据扩散系数,我们可以计算出给出这些1h峰的物种的流体力学直径。存在3个dh值,其可被分配为来自纳米颗粒和两种类型的游离水解硅烷的dh。因此,可以从该频谱中得出一些结论。首先,两个预期的官能团已经很好地接枝在颗粒上。第二,纯化的颗粒具有稳定的接枝功能,而具有非常少的游离水解的硅烷。最后,该颗粒的流体动力学直径约为5.0nm,这与dls测得的结果一致。通过将仅来自cest的1h峰的面积与来自cest和taned的所有1h峰的总面积进行比较,可以计算出样品中cest量与taned量之间的比率。在此实施例中,结果为cest/taned=1.30。表1和图5示出了固态29sinmr光谱的结果。首先,45%的si(t2和t3)来自有机硅烷,即taned和cest。通过将该值与通过比较1h峰面积得到的结果相结合,我们可以计算出25.45%来自cest以及19.55%来自taned。然后剩余的55%的si(q2、q3和q4)来自teos。表1根据以上结果,我们可以按如下taned:cest:teos=1.00∶1.30∶2.81建立所有组合物的摩尔比。此外,我们还可以根据该比率计算出taned的含量,约为0.784μmol/mg。taned的含量也可以以ebt(或net)作为颜色指示剂通过比色法进行定量。将冻干粉末再分散在水中以获得48mg/ml溶液(a)。将100μla、作为颜色指示剂的10μlebt和10ml氨水缓冲液的溶液用5mmcacl2溶液滴定。在此实施例中,结果约为0.855μmol/mg。实施例2:使用大环-螯合剂-官能化硅烷(aptes-dotaga)和氨基硅烷(aptes)在水中合成超小杂化螯合二氧化硅纳米颗粒(uchsnp)在此实施例中,合成分为两个步骤。首先,合成尚未商业生产的硅烷前体aptes-dotaga。然后,根据实施例1中所述的方法合成用大环螯合剂dotaga官能化的超小杂化螯合二氧化硅纳米颗粒(uchsnp)。步骤1:合成大环-螯合剂-官能化硅烷(aptes-dotaga)aptes-dotaga可通过两种不同的方法合成:用hbtu(2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基铀六氟磷酸盐)通过aptes与丁基保护的dotaga上的活化羧基反应,步骤1a或通过aptes与dotaga酸酐反应,步骤1b。步骤1a:由丁基保护的dotaga合成大环-螯合剂-官能化硅烷(aptes-dotaga)可从叔丁基保护的dotaga合成aptes-dotaga,该dotaga通过肽偶联与aptes偶联。然后,将中间体脱保护,得到最终化合物。反应图式如图6所示。将1g的(t-bu)4dotaga、0.6g的hbtu和0.2g的hobt称重到100ml的圆底烧瓶中,向该烧瓶中加入28ml的dcm(二氯甲烷),然后添加1.3ml的dipea。将混合物搅拌15min,然后将0.3gaptes注射到反应混合物中。将该溶液在室温下搅拌过夜。使用dcm将反应混合物稀释三次至90ml,然后在分液漏斗中用80-90ml柠檬酸溶液(ph3)萃取反应溶液。分离的有机相进一步用80-90ml5%w/vnahco3萃取,最后用蒸馏水萃取。上述提取能够去除偶联剂/未反应的aptes/外部水溶性成分。分离的有机相经mgso4(5g)干燥5min,并连续过滤以得到澄清的滤液。使用rotavapor在30℃下蒸发滤液,得到浅褐色粘性残余物(中间体)。使用hrms验证了中间体的形成。c44h85n5o12si的m/z:计算值:926.5856,获得值:926.5849(m+na)+。1hnmr(500mhz,cdcl3)δ0.4–0.7(m,2h),0.7–0.8(m,1h),1.0(dd,j=9.0,6.7hz,1h),1.1–1.2(m,9h),1.3–1.5(m,32h),1.5(p,j=7.8hz,2h),1.7(d,1h),1.9–2.1(m,1h),2.1–2.3(m,1h),2.4–3.4(m,29h),3.5–3.7(m,1h),3.7–3.8(m,4h)。13cnmr(126mhz,cdcl3)δ7.5,7.8,18.3,20.4,23.5,25.9,26.8,27.8,27.8,27.9,27.9,27.9,28.2,28.3,29.7,33.0,38.6,42.1,47.6,49.8,58.4,63.6,80.8,82.3,171.1,173.2。将上述残余物与20ml浓hcl混合并搅拌40min,然后使用旋转蒸发仪在35℃去除过量的酸,得到固体残余物。将残余物进一步溶于约10ml水中,并进行旋转蒸发以去除游离酸。将获得的浓缩物溶于10ml水中,并立即在氮气中冷冻并冻干,得到1.11g浅棕色粉末。c22h41n5o12si的m/z:计算值:618.2413,获得值:618.2425(m+na)+。1hnmr(500mhz,deuteriumoxide)δ0.5–0.8(m,2h),1.2–1.3(m,1h),1.4–1.6(m,1h),1.6–1.8(m,1h),1.8–2.2(m,1h),2.3–4.5(m,26h)。步骤1b:由dotaga酸酐合成大环-螯合剂-官能化硅烷(aptes-dotaga)硅烷前体aptes-dotaga也可由dotaga酸酐合成。反应图式如图7所示。在典型的实施例中,将9.375g(16.36mmol)的dotaga酸酐置于1l的圆底烧瓶中。然后,快速添加494ml无水dmso和1.933ml(8.16mmol)aptes。使用过量dotaga酸酐以确保所有aptes都反应。这允许在下一步中精确控制最终颗粒的组成。将反应置于氩气气氛下,在15-20h内加热至75℃。产物形成白色沉淀。将产物转移至5l丙酮中并在4℃下保持48h,使其完全沉淀。沉淀物通过42号滤纸过滤。使用约2-3l丙酮洗涤沉淀物以去除dmso。通过在37℃蒸发过夜去除残留的丙酮。纯化后,将沉淀物溶于水中,并根据方法2进行hplc分析。通过将合成混合物的色谱图与gdcl3以及gdcl3和dotaga(gd3+)的混合物的色谱图叠加,可以鉴定峰。为了证实反应产物的结构,还进行了ms分析。ms光谱在594、475、296.5和237m/z处显示4个主峰,与提议产品的模拟光谱相符,这表明单电荷离子和双电荷离子的共同存在。通过比色法和eu磷光滴定测定的总aptes-dotaga和未反应dotaga的含量以及根据方法3的色谱法的未反应dotaga的含量间接测定合成混合物中aptes-dotaga的含量(mol/g)。对于比色法,将200μl8.815mg/l的合成混合物和25μlebt(net)添加到10mlph10的氨水缓冲液中。用5mm的cacl2滴定该溶液。终点体积为600μl,使得总dotaga含量为1.70μmol/mg。利用该结果,在ph5的醋酸盐缓冲液中制备了一系列44.075mg/l的合成混合物的样品,并增加了eu3+(0μm–140μm)的浓度,以更精确地测定总dotaga的含量。在进行磷光测量之前,将这些样品在80℃下温育48h。图8-a示出了dotaga(eu3+)在40μm处的发射光谱。在594nm和616nm处发现两个发射峰。图8-b分别示出了在594nm处(圆形)和616nm处(方形)的滴定曲线。dotaga的总含量测定为1.475μmol/mg。因为比色法由于难以识别颜色开始变化的点而趋向于高估结果,因此该结果更为精确。使用hplc分析和校准曲线,计算dotaga(gd3+)的浓度。从该结果推导出未反应的dotaga的含量。未反应的dotaga的计算含量为0.745μmol/mg。从以上结果可以推断出预期产物aptes-dotaga的含量为0.730μmol/mg。这表明几乎所有aptes都与过量的dotaga反应以形成aptes-dotaga,且混合物中没有aptes残留。计算表明,反应的收率为99%,整个过程(即反应和过滤)的分离收率为79%。还使用红外(ir)光谱表征aptes-dotaga。将dotaga以及aptes-dotaga的反应混合物溶于水中并调节至ph2以使羧基质子化。这将c=o酰胺在1677cm-1处的峰与c=o羧基在1713cm-1处的峰区分开。将两种溶液在80℃下干燥4天。用干燥粉末获得红外光谱。图8c示出了dotaga粉末和aptes-dotaga反应混合物的红外光谱。下表2示出了一些重要峰的分布。在1677.0cm-1处出现峰,在1712.7cm-1处出现峰强度降低,这表明形成了酰胺键。表2.dotaga和aptes-dotaga红外光谱中主要峰的分配波数(cm-1)分配3385.3si-oh拉伸或伯胺n-h拉伸3238.7si-oh拉伸或仲酰胺n-h拉伸或羧基o-h拉伸3079.2仲酰胺ii泛音或羧基o-h拉伸2932.6亚甲基不对称c-h拉伸2885.2亚甲基对称c-h拉伸1712.7羧酸c=o拉伸1677.0仲酰胺c=o拉伸1622.2胺nh2剪式,n-h弯曲1385.1羧酸c-o-h面内弯曲1221.3羧酸c-o拉伸或脂族c-n拉伸1122.1si-o-si不对称拉伸或脂族c-n拉伸1087.6si-o-c拉伸步骤2:由aptes-dotaga合成uchsnp可以采用两种策略由aptes-dotaga合成uchsnp:a)可以从一开始就直接使用aptes-dotaga硅烷来合成表面上带有游离螯合剂的颗粒(uchsnp),然后将这些uchsnp与gd3+络合以形成最终产物(uchsnp@gd-1);或b)可以在水解-缩合过程之前将aptes-dotaga硅烷与gd3+络合,以产生具有络合螯合物的最终颗粒(uchsnp@gd-2)。图9总结了这两种策略。步骤2a:从空位aptes-dotaga合成uchsnp@gd-1将200ml水添加到由步骤1a或1b合成的产物中,在任一情况下,均含2.228mmol的aptes-dotaga。通过添加适当浓度的naoh溶液将溶液的ph值从约3调节至9。将溶液搅拌1-2h,以将aptes-dotaga溶解并释放为单体形式。然后,将teos(1015.4μl,4.457mmol)和aptes(526.7μl,2.228mmol)逐一添加到上述溶液中。如有必要,应将ph值调回9,因为添加aptes会略微增加ph值。添加水以使aptes-dotaga、teos和aptes的最终浓度分别为10mm、20mm和10mm。将反应混合物在25℃下搅拌过夜,以使溶液变得均匀,这意味着所有乙氧基硅烷均已水解。然后,通过添加几滴适当浓度的hcl将ph从9降至7。将溶液在25℃下搅拌2h,然后将其从ph7重新调整为ph4.5。将溶液在25℃下再搅拌1-2h,然后加热至80℃并在油浴中缓和搅拌过夜(15-20h)。将少量溶液(1ml)通过0.2μm膜过滤,并通过dls和hplc分析(根据方法1)。对于hplc分析,制备了两种类型的样品。首先,将过滤溶液快速稀释2倍,使其在注入hplc系统之前具有5g/l的理论浓度。在295nm处跟踪信号,这是aguix颗粒的典型吸收。其次,将200μl的过滤溶液与5μl的506mmcuso4混合,其与溶液中螯合剂的理论浓度相比过量。将该溶液在80℃下温育2h。确认温育后的ph并保持在3.23,该ph足够低以避免形成cu(oh)2。然后,将溶液迅速稀释至5g/l的理论浓度。在700nm处跟踪信号,这是铜及其dotaga络合物的特定吸收峰。通过vivaspintm(mwco=3kda)经超滤将溶液浓缩至10ml。再次,重复hplc分析。将不具有铜和铜络合的2个样品就在注入hplc系统之前稀释至5g/l的理论浓度,以与之前的结果进行比较。然后,将该溶液进一步用超滤纯化。如果前体是与未反应dotaga的混合物,则在纯化前应通过添加hcl溶液将溶液的ph值调节至2。该步骤使dotaga去质子化,并使它们不再静电附着于新形成的颗粒表面上的氨基。将溶液离心直至剩余一半的体积(纯化速率21=2)。通过向试管中填充10-4m的盐酸(hcl)溶液(或如果在ph2进行过滤,则添加10-2m的hcl溶液)并再次离心,直至从hplc色谱图计算出的纯度达到≥90%(通常,210=1024纯化速率)。然后,将溶液通过0.2μm的膜过滤以去除灰尘。最后,将溶液冻干以长期保存。获得706mg的冻干粉末。图10示出了已形成的uchsnp-1的dh分布。颗粒的dh约为4.6±1.6nm。hplc分析证实,纯化后,纳米颗粒是纯的并在表面上很好地接枝了dotaga。后者通过700nm的色谱图表征。通过两种方法对最终纳米颗粒(uchsnp-1)中dotaga的含量进行定量:1)铜hplc分析(使用方法1)和2)eu磷光滴定。对于第一种方法,在ph3下制备含142.8g/luchsnp-1和200mmcuso4的混合物,并在80℃下温育2h。就在注入hplc系统之前,将溶液在10-3m的hcl溶液中稀释20倍。hplc色谱图显示纳入金属的纳米颗粒和游离铜络合的aptes-dotaga的存在。纳入金属的纳米颗粒的rt比初始空位纳米颗粒长。这可以通过络合引起的表面电荷或电离态的变化来解释。更重要的是,纳米颗粒峰的形状在络合后显示出均匀的分布。所有铜物种的总浓度为10.03mm,精确等于理论上引入的量。从这些结果中,我们可以测定样品中dotaga的总浓度并推断其含量约为0.72μmol/mg。为了通过eu进行滴定,在ph5的醋酸盐缓冲液中制备了uchsnp-1的浓度为44.08mg/l和eucl3的浓度为0μm-140μm的一系列样品。在测量之前,将这些样品在80℃下温育48h。图11显示了dotaga(eu3+)络合物在2个特定发射波长的滴定曲线。结果为0.79μmol/mg。uchsnp@lu-1为了评估uchsnp-1的直径、其表面上aptes-dotaga和aptes的存在及其比率,从空位uchsnp-1和与抗磁性镧系元素离子(即lu3+)络合的uchsnp-1(uchsnp@lu-1)收集1hnmr和nmrdosy光谱。对于空位uchsnp-1样品,将冻干粉末再分散在水中。将溶液的ph值调节至7.4,然后添加水,以在dotaga中具有127g/l或100mm的最终浓度。将溶液冻干并以相同浓度再分散在d2o中。然后,将470-500μl样品添加到nmr管中进行测量。对于uchsnp@lu-1样品,将冻干粉末再分散在水中。使用由eu滴定法计算得到的dotaga含量,分4次缓慢添加浓度为1.98m的32.5μl的lucl3溶液(摩尔比dotaga:lu=1:0.9)。在每次之间,通过添加适当浓度的naoh溶液将ph小心地提高至4-5,然后再添加下一个。添加4次后,ph为5。将该溶液在80℃下温育48h。最后,将ph值提高到7.4,并添加水以在dotaga中具有127g/l或100mm的最终浓度。将溶液冻干并以相同浓度再分散在d2o中。然后,将470-500μl样品添加到nmr管中进行测量。图12示出了uchsnp-1的nmr光谱。首先,图12-a示出了d2o中的2dnmrdosy光谱。大多数质子似乎具有相同的54.4μm2/s的扩散系数(d)。结果表明预期的官能团(即aptes-dotaga和aptes)被良好地接枝在相同的颗粒上。其次,有一些游离的水解硅烷具有更快的系数(194.5μm2/s)。发现uchsnp-1@lu的光谱相似(图12-c)。主要产品的仍具有约为56μm2/s的d值。它们与其他一些具有更快d值(410和214μm2/s)的较小物种共存。可以根据爱因斯坦方程计算主要颗粒的流体动力学直径,对于uchsnp-1和uchsnp-1@lu而言,其分别约为7.0nm和6.8nm。溶液的粘度未知,且在该浓度下可能远高于纯d2o。因而,计算的dh可能会被高估。在任何情况下,它都保持在10nm以下,并且与dls测得的结果一致。图12-b和d示出了uchsnp-1和uchsnp-1@lu的1d1h光谱的峰积分。归因于可在其他地方得到证实的dotaga的复杂1h谱,大多数1h峰重叠。幸运的是,dotaga在小于1ppm的区域没有1h峰,在该位置可发现最接近aptes和aptes-dotaga的si的1位碳的1h峰(图12-e)。因而,通过将来自那些碳1的1h峰的面积与所有1h峰的总面积进行比较,我们可以测定样品中aptes量与aptes-dotaga量之间的比率。在这两个样品中,结果分别为1.35和1.12。uchsnp@gd-1为了使uchsnp-1具有mri显影增强以及放射增敏性能,将gd3+络合在颗粒上。在一个典型实施例中,将333mguchsnp-1的冻干粉末再分散在水中。分三次缓慢添加36μl2.188m的gdcl3溶液(dotaga:gd的摩尔比=1:0.9)。在每次之间,通过添加适当浓度的naoh溶液将ph小心地提高至4-5,然后再添加下一个。添加3次后,ph为5。将该溶液在80℃下温育48h。温育后,保持ph。通过切向过滤(mwco=3kda)以纯化速率5纯化该溶液,以去除任何游离的gd3+。通过hplc评价溶液的纯度(方法1)。用hcl10-2m溶液将纯化溶液(~1ml)稀释52倍以使其就在注入hplc系统之前,在dotaga中具有5mm的理论浓度。纳入gd3+的纳米颗粒的rt比纳入cu2+的纳米颗粒和初始空位纳米颗粒长。这可以再次通过由络合物引起的表面电荷或电离态的变化来解释,因为dotaga(gd3+)具有与金属配位的所有四个羧酸盐基团,而dotaga(cu2+)具有两个游离羧酸盐基团。更重要的是,如在纳入铜的纳米颗粒的情况下,纳米颗粒峰的形状在络合后显示出均匀的分布。hplc色谱图显示该纳米颗粒是纯的。从在295nm处的色谱图评估溶液的纯度,其接近100%。接下来,将溶液的ph提高至7.4,并且将溶液通过0.2μm膜过滤以在冻干之前去除灰尘。在该实施例中,获得了250mguchsnp@gd-1粉末。将冻干粉末样品再分散在水中,以验证流体动力学直径(dh)、表面电荷和弛豫度(r1和r2)。图13示出了5.7±1.3nm的uchsnp@gd-1的dh的分布直方图。表面电荷由zeta电位值反映,该zeta电位值在ph6.65下为-5.8mv,且在ph7.34下为-8.2mv。然后,进行元素分析以揭示颗粒的化学组成。根据该结果,gd的含量为0.60μmol/mg(表3)。接下来,测量100g/luchsnp@gd-1的水溶液(60.4mmgd)的弛豫时间(t1和t2),以在37℃和1.4t下得出r1=21.4(s-1.mm-1)和r2=34.1(s-1.mm-1)。步骤2b:由络合的aptes-dotaga(gd3+)合成uchsnp@gd-2将200ml水添加到2.333mmol由步骤1a或1b合成的aptes-dotaga中。通过添加适当浓度的naoh溶液将溶液的ph调节至4。分3次添加1.938ml的2.188m的gdcl3溶液(摩尔比(aptes-dotaga+dotaga):gd=1∶0.9)。在每次之间,通过添加适当浓度的naoh溶液将ph小心地提高至4-5,然后再添加下一个。添加3次后,ph为5。将该溶液在80℃下温育。验证ph值,每24h后将其重新调节至5。温育48h后,ph稳定地保持在5。然后,将该溶液的ph调节至9,并将溶液搅拌1-2h,以将aptes-dotaga(gd3+)溶解并释放成单体形式。然后,将teos(1015.4μl,4.457mmol)和aptes(526.7μl,2.228mmol)逐一添加到上述溶液中。如有必要,应将ph值调回9,因为添加aptes会略微增加ph值。添加水以使aptes-dotaga(gd3+)、aptes-dotaga、teos和aptes的最终理论浓度分别达到9mm、20mm和10mm。将反应混合物在25℃下搅拌过夜,以使溶液变得均匀,这意味着所有乙氧基硅烷均已水解。然后,通过添加几滴适当浓度的hcl将ph从9降至7。将溶液在25℃下搅拌2h,然后将其从ph7重新调节至ph4.5。将溶液在25℃下再搅拌1-2h,然后加热至80℃并在油浴中缓和搅拌过夜(15-20h)。将少量溶液通过0.2μm膜过滤,并通过dls和hplc分析(根据方法1)。为了进行hplc分析,将过滤溶液快速稀释两倍,以就在注入hplc系统之前具有5g/l的理论浓度。在295nm处跟踪信号。此外,还使用荧光检测器(λex=274nm,λem=312nm)定性检测gd络合物的存在。然后,通过vivaspintm(mwco=3kda)将溶液浓缩至10ml。再次,重复hplc分析。就在将样品注入hplc系统之前,将样品稀释至5g/l的理论浓度,以与之前的结果进行比较。然后,进一步用超滤将该溶液纯化。如果前体是与未反应dotaga的混合物,则在纯化前应通过添加hcl溶液将溶液的ph值调节至2。进行纯化,直到从hplc色谱图计算出的纯度达到≥90%(纯化速率10)。然后,在冻干之前将溶液的ph提高至7.4。将其通过0.2μm膜过滤以去除灰尘和大颗粒,然后将其冻干以长期保存。在该实施例中,获得了716mguchsnp@gd-2粉末。图14示出了已形成的uchsnp@gd-2的dh分布。颗粒的dh约为3.9±1.3nm。hplc色谱图表明,纯化后,纳米颗粒是纯的并在表面上良好地接枝有dotaga。络合后,峰的形状显示出均匀的分布。由295nm处的色谱图评估溶液的纯度,其为96.8%。将冻干粉末样品再分散在水中,以验证冻干后的dh、表面电荷和弛豫度(r1和r2)。图15示出了2.8±0.7nm的uchsnp@gd-2的dh的分布直方图。表面电荷由zeta电位值反映,其在ph7.4时,为–35.6mv。然后,还进行了元素分析以揭示颗粒的化学组成。根据该结果,gd的含量为μmol/mg(表3)。根据从该结果推断的组成比率,uchsnp@gd-2在其结构中具有更少的aptes和更多的游离dotaga,这解释了具有这些纳米颗粒所测得的负zeta电位更大。uchsnp@gd-2上游离dotaga的含量通过另一次eu滴定进行验证(图16)。结果表明,在总含量为0.94μmol/mgdotaga中,至少有0.20μmol/mg或24.1%的dotaga可有效地与金属反应。这高于uchsnp@gd-1的情况(10%)。元素分析表明,aptes-dotaga的总含量总是比滴定法估计的值高10%,这表明两种纳米颗粒中的某些dotaga是金属无法接近的。uchsnp@gd-2中较高含量的游离dotaga可能归因于分解,当将反应混合物在ph9下搅拌过夜以水解teos时,所述分解加速。与uchsnp@gd-1相比,uchsnp@gd-2中aptes的量较低,这可以用aptes-dotaga和aptes-dotaga(gd3+)电荷的差异来解释。在ph4.5下,当络合形式始终为-1时,dotaga的游离形式为-2或-3。也许,该络合形式具有较弱的电荷排斥力,这使其比游离形式更容易与氨基硅烷竞争,从而在teos产生的聚硅氧烷表面上获得位置。接下来,测量100g/luchsnp@gd-2的水溶液(63.0mmgd)的弛豫时间(t1和t2),以在37℃和1.4t下得出r1=18.5(s-1.mm-1)和r2=28.7(s-1.mm-1)。表4总结了uchsnp、uchsnp@gd-1和uchsnp@gd-2的特性和性能。表3纳米颗粒的元素分析表4.uchsnp、uchsnp@gd-1和uchsnp@gd-2的特性和性能*a-d:aptes-dotaga,a:aptes,t:teos,实施例3:合成不同尺寸uchsnp通过将合成aptes-dotaga与合成聚硅氧烷颗粒合并为一锅法方案,可以进一步简化合成。此外,可以通过改变式中硅烷前体的比率来控制颗粒尺寸(图17)。将8gdotaga酸酐(13.96mmoles)置于100ml圆底烧瓶中,向其中快速添加31.6ml无水dmso和3.300mlaptes(13.96mmoles)。在氩气气氛下搅拌反应,并在15-20h内加热至75℃。与第二个实施例不同,由于过量aptes的存在会电离dotaga的羧基,因此产物可溶。将混合物冷却至室温,然后添加663ml超纯水。dmso在溶剂中的最终百分比应小于5%,以免溶解下一步骤中使用的切向过滤膜。通过之前引人的通过gd3+探测的eu滴定和hplc的组合,取少量样品以定量产生的aptes-dotaga的量。通过添加naoh溶液将溶液的ph调节至9,并将混合物搅拌1h以充分释放单体形式的aptes-dotaga。然后,将溶液分成4个体积。如表5所示,将增加量的teos和水添加到每个体积中,以确保所有样品中的总硅烷浓度为20mm。验证最终溶液的ph,并在必要时将其重新调节至9。将这些溶液在25℃搅拌过夜。表6示出了所用的不同硅烷之间的比率a、b和c。表5.实施例2中teos、h2o的添加量和不同配方的组成表6:比率a、b和c对于未添加teos的uchsnp-3样品,在暴露于碱性ph过夜后,取少量样品以验证aptes-dotaga的量。图18示出了两个样品的eu滴定结果:暴露于碱性ph过夜之前(a)和之后(b)。还进行了hplc分析。表7总结了这些测试的结果。表7.总dotaga和未反应dotaga浓度的结果汇总显然,暴露于ph9不会影响dotaga结构或aptes-dotaga的酰胺键。dotaga峰之间的微小变化可能是由于室温的差异所致。最终,根据该结果,大约70%的dotaga酸酐已经反应。如此,我们可以重新计算4个纳米颗粒样品中组成的精确含量,如表5所示。第二天,将4种溶液的ph重新调节至7。将它们搅拌1h,然后将其ph重新调节至4.5。将溶液再搅拌一小时,然后将其加热至40℃过夜。取1ml这些溶液用于dls测量(图19-a)。来自uchsnp-3的信号太弱,无法获得可靠的值,这意味着会生成非常小的纳米颗粒和/或稀释浓度的纳米颗粒。其他3个样品显示出纳米颗粒尺寸对teos添加量的明显依赖性。uchsnp-4、5和6的dh分别为5.2、7.5和13.6nm。通过vivaspin(mwco=3kda)将这些溶液浓缩至合适的体积,其中aptes-dotaga的理论浓度达到200mm。然后,将溶液的ph值调节至2,以将未反应的dotaga和aptes-dotaga从它们与新形成的纳米颗粒表面上的胺的静电相互作用中释放出来。使用10-2mhcl作为洗涤溶剂,通过vivaspin在此ph值下以纯化速率64纯化溶液。就在注入hplc之前,将每种纯化溶液的少量样品在10-2mhcl中稀释40倍,以在295nm处进行紫外吸收分析。配方中添加的teos越多,纳米颗粒的滞留时间越慢。这间接表明纳米颗粒尺寸与teos量有关,因为更高的tr通常意味着更大的纳米颗粒。数据总结于表8。表8.不同比率起始硅烷的纳米颗粒的色谱结果总结纯化后,溶液通过0.2μm膜过滤以去除灰尘和其他大颗粒,然后冻干以长期保存。将少量的4个样品以100g/l再分散在水中。由于在冻干前未调节溶液的ph,因此在再分散后,其ph保持为约2。uchsnp-6粉末无法在该ph值下再次分散在水中。在dls中进行测量之前,用10-2m的hcl将这些母液快速稀释至10g/l。将另一系列的样品以150g/l再分散。添加naoh溶液以中和样品至ph7。在这种条件下,可以再分散uchsnp-6。根据样品的不同,决定是否添加水以获得约80-100g/l的最终浓度。同样,用水将这些母液快速稀释至10g/l,然后再在dls中进行测量。图19-b示出了在ph2下3个样品uchsnp-3、4、5的流体动力学直径分布。同时,图19-c示出了在ph7下获得的4个样品的流体动力学直径分布。它们一起清楚地表明配方中teos增加时,np尺寸增加。数据总结于表9。表9.不同比率起始硅烷的nps的dls结果总结uchsnp-6不能在ph2下再分散的事实可能是由于其较低的胶体稳定性,这可以通过较大的尺寸、较高的teos比率以及在其周围的aptes-dotaga和aptes保护层的较低密度来解释。值得一提的是,在此ph下,dotaga的4个羧基被质子化。因而,颗粒之间的排斥仅依赖于aptes氨基(但其非常短)的正电荷以及dotaga的阻碍作用。这就解释了为什么在ph7下uchsnp-6可以毫无问题地再分散。在这种情况下,dotaga的2个羧基被去质子化并提高了总排斥力。ph7下的结果与纯化前的值非常相似(图19-a),这意味着该过程中大多数颗粒保持完整。ph7下的直径略大于ph2下的直径。或许,这是由于每个颗粒上去质子化的dotaga之间的排斥,使得它们更伸展了。简而言之,该结果表明,通过改变硅氧烷网络生成剂、teos、和有机硅烷、aptes-dotaga和aptes的比率,我们可以控制np的尺寸。在该实施例中,teos∶(aptes-dotaga+aptes)的比率为1:1,纳米颗粒尺寸超过6nm,这是快速肾脏清除的推荐限值。在1.5:1的比率下,其尺寸将超过10nm,更具体地达到14nm。如图20所示,通过eu滴定测定每个样品的dotaga含量。我们清楚地看到,当配方中的teos如预期增加时,dotaga含量下降。有趣的是,我们投入的teos越多,最终aptes-dotaga的产量就越高。据推测,硅酸和有机硅烷醇之间的硅氧烷键强于有机硅烷醇自身之间的硅氧烷键。另外,已知氨基硅烷,特别是具有短碳链的氨基硅烷(诸如aptes),具有相当低的水解稳定性。也通过弛豫法和元素分析表征了纳米颗粒uchsnp-3、uchsnp-4、uchsnp-5和uchsnp-6。表10总结了uchsnp-3、uchsnp-4、uchsnp-5和uchsnp-6的性能和特性。表10.uchsnp-3、uchsnp-4、uchsnp-5和uchsnp-6的特性和性能*a-d:aptes-dotaga,a:aptes,t:teos,实施例4:在二甘醇中,采用一锅法中等规模地合成uchsnp(uchsnp-7)及其与不同金属的络合物将6.187mlaptes(26.17mmol)添加到一个装有90ml二甘醇(deg)的200ml玻璃瓶中。将溶液在室温搅拌1h,然后添加10gdotaga酸酐(17.45mmol)。将混合物在室温搅拌6天,以使反应完全。该产物是细悬浮液。取出少量样品,并在水中稀释10倍以测量dls中的dh。将7.952ml的teos(34.90mmol)添加到悬浮液中。将该混合物搅拌1h。取出少量样品,并在水中稀释10倍以测量dls中的dh。然后,添加900ml超纯水。溶剂中deg的最终百分比应小于10%,以免溶解下一步骤中使用的切向过滤膜。将该混合物加热至50℃并保持搅拌18h以使teos完全水解。取少量样品以分别通过dls和hplc分析新形成的颗粒的流体动力学尺寸和色谱图。通过vivaflow盒(mwco=5kda)将溶液浓缩至200ml。然后,将溶液的ph调节至2以破坏未反应的dotaga和aptes-dotaga与新形成的纳米颗粒表面上的胺之间的离子相互作用。使用vivaflow在此ph值下以水作为洗涤溶剂(200ml-1l-200ml-1l-100ml)以纯化速率50纯化溶液。纯化后,通过滴加1mnaoh溶液将溶液中和至ph7.4,并通过0.2μm膜过滤以去除灰尘和其他大颗粒,然后冻干以长期保存。先将少量的纯化溶液样品在水中稀释10倍,然后再通过dls进行分析,或者先在0.1%的tfa水溶液中稀释10倍,然后使用295nm处的uv吸收通过hplc进行分析。图21-a示出了合成过程中不同步骤的dls图:aptes和dotaga酸酐在deg中的混合物(虚线,方形)、添加teos后的相同混合物(虚线,上三角)、在水中稀释并加热过夜后的混合物(虚线,下三角)、通过0.2μm膜过滤后的相同混合物(实线,圆形)。图21-b示出了通过vivaflow纯化后的颗粒的dls图。首先,用水将在deg中的样品稀释10倍,然后过滤以获得均质溶液,而不是初始悬浮液。如表11中总结的,uchsnp-7的dh在合成过程中改变。在添加teos之前,没有产生任何颗粒,这表明2个第一样品(0.9nm和1.1nm)的dh值较小。引入teos并水解后,它开始产生超小的核,其他有机硅烷(即aptes和aptes-dotaga)现在可以更稳定地接枝在其上。最终颗粒的dh约为4nm。表11.合成过程中不同步骤的uchsnp-7的dls结果总结纯化后,颗粒具有几乎相同的tr且峰宽略小,这可以简单解释为去除了物理吸附的硅烷。纯度达到98%。从纯化后的颗粒的峰面积与纯化前的颗粒和反应物的总峰面积之比可以粗略估计反应和纯化的收率,其约为40%。为获得更多的定量分析,数据总结在表12中。表12.纯化前、后uchsnp-7的hplc结果总结通过eu滴定测定uchsnp-7的dotaga含量,如图22所示。结果(~0.8μmol/mg)与先前实施例中具有相似dh的颗粒中发现的结果一致。将该结果与所生产颗粒的总重量(5.388g)结合,我们可以测定该工艺的收率,其与引入的dotaga的量相比,约为24.7%。也通过nmr、zeta电位法、弛豫法和元素分析表征纳米颗粒uchsnp-7。表13总结了uchsnp-7的性能和特性,且表14示出了在不同ph下uchsnp-7的zeta电位。uchsnp-7在不同ph下的zeta电位的完整曲线如图24a所示。表13.uchsnp-7的特性和性能*a-d:aptes-dotaga,a:aptes,t:teos表14.在不同ph下空位uchsnp-7的zata电位将uchsnp-7与不同金属(gd、ho、tb和bi)络合将283mg含227μmoldotaga的uchsnp-7的冻干粉末再分散在水中,以使dotaga的浓度约为200mm。通过添加适当浓度的naoh溶液将溶液的ph调节至5.5。分三次缓慢添加98.5μl2.188m的gdcl3溶液(摩尔比dotaga:gd=1:0.95),同时在70℃的加热板上加热并搅拌该溶液以加速络合。在每次之间,通过缓慢添加naoh溶液将ph小心地提高至5-5.5,然后再添加下一个。添加3次后,添加水以获得浓度为100mm的dotaga且ph约为5-5.5。将该溶液在80℃的油浴中搅拌18h。温育后,ph保持在约5.5。用水作为溶剂通过切向过滤(mwco=3kda)以纯化速率16将该溶液纯化,以去除任何游离的gd3+。最后,通过添加几滴naoh溶液将溶液中和至ph7并通过0.2μm膜过滤,以去除灰尘和其他大颗粒,然后进行冻干以进行长期保存。将少量纯化溶液样品在水中稀释10倍,然后再通过dls分析。使用431μl500mm的hocl3或tbcl3溶液的替代方案应用了相似的方案。对于bi颗粒,由于氢氧化铋的溶解度非常有限,因此使用了817μl在6mhcl中的250mmbicl3溶液。在进行任何添加以增加bi3+的溶解度和络合速度之前,必须将纳米颗粒溶液加热至70℃。未能保持该条件可能会导致氢氧化铋沉淀的形成。在每次添加步骤之间,需要使用10m的naoh溶液将溶液中和至ph5-5.5,并将溶液在80℃的油浴中加热1h。该方案的其余部分类似。图23示出了uchsnp-7@m(m:gd、tb、ho或bi)的dls图。结果定量地呈现在表15中。所有4个颗粒的流体力学直径均约为6nm。该结果与实施例1一致,其中uchsnp@gd-1也具有约6nm的dh。表15.uchsnp-7@m的dls结果总结将bi颗粒的纯化溶液(uchsnp-7@bi)在0.1%tfa水溶液中稀释15倍,然后通过hplc分析(方法1)。在tr=15.7min处发现了纳米颗粒的峰,这与uchsnp@gd-1非常相似。峰的形状也显示出络合后的均匀分布。颗粒的纯度非常高(97.4%)。将此色谱图归一化为与uchsnp-7相同的高度,以比较滞留时间(tr)和峰宽(fwhm)。络合颗粒的2个值均高于空位颗粒的值。结果总结在表16中。表16.与bi络合之前和之后的uchsnp-7的hplc结果总结也通过zeta电位法、弛豫法、icp-eos、紫外可见光谱和红外光谱表征纳米颗粒uchsnp-7@gd、uchsnp-7@tb、uchsnp-7@ho和uchsnp-7@bi。表17总结了uchsnp-7的性能和特性。图24b示出了在ph6.6下纳米颗粒的zeta电势。纳米颗粒的紫外可见光谱如图25所示。uchsnp-7@bi样品在309nm处显示出强峰,这是dota(bi3+)络合物的典型峰。uchsnp-7@ho的紫外可见光谱显示出ho3+的几个吸收峰。红外光谱如图26所示。它们证实了金属的存在。表17.金属络合uchsnp-7的性能实施例5:体内磁共振成像(mri)实验将三只balb/c小鼠的两侧皮下接种结肠癌(ct26)细胞。将uchsnp@gd-2冻干粉末以100mm(以gd计)分散在生理血清中。将该浓缩溶液在血清中稀释至20mm,然后以每公斤200μmol(以gd计)的剂量向小鼠静脉内注射。使用带有1h40mm线圈、paravision5.11软件的7tmri系统300wb微成像光谱仪(bruvision,德国)在注射之前(对比前)和注射之后(对比后)采集图像。通过调节异氟烷浓度(1.5%)连续监测呼吸频率。使用intragateflash多层切片记录动态对比度增强(dce)序列在tr=100ms、te=4ms、flipangle=80°下的无运动伪像(motionfreeartifacts)。重复次数设定为15,要重建的时间范围为1。选择3厘米x3厘米的视野(fov)和256x256矩阵的厚度为1mm的4个切片,以在平面上提供117μmx117μm的空间分辨率。总扫描时间约为3分14秒。最后,将intragateflash多层切片序列的细长版本用于动态跟踪,以在40min的扫描时间内获得相同的瞬时分辨率。3-6小时对比后进行2-3min的扫描作为跟踪。监测肿瘤和肝中的几个目标区域(roi),并在颗粒注射前和注射后绘制roi的mri强度。计算每个组织区域中信号的组织增强水平(st–s0)/s0,其中st是注射后每个时间点测量的信号强度,s0是注射前的信号强度。图27a示出了mri横截面,其中如所期望的突出了肿瘤区域。对比前和对比后的图像比较清楚地显示由颗粒引起的肿瘤区域的较高亮度。对比增强表示为与对比前图像相比增强的百分比。在肿瘤组织中,uchsnp@gd-2显示了在注射后30分钟达到最大增强(35%的信号增强)的摄入期以及具有3小时半衰期的延长的清除期,这证明了epr效果(增强的渗透性和滞留性)(参见图27b)。在肝中,在注射颗粒后6分钟观察到增强峰(信号增强90%),然后进入清除阶段(参见图27c)。注射后40分钟后,信号强度为最大强度的一半,表明肝半衰期为约30分钟。在通过肝中的血管网络进行补充性短暂可视化的血液循环后,如先前使用超小纳米颗粒所示,颗粒从肾脏皮层排到膀胱。这项影像学研究证明,在整个观察期内,uchsnp@gd-2在肿瘤和肝组织中均显示对比度增强,而没有观察到典型的大分子药剂肝积聚。因此,它们改善了成像性能而没有不希望的肝摄取。同时,在肿瘤中相对较长的滞留时间为向肿瘤组织的向量化打开了前景。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1