阿哌沙班药物组合物及其制备方法与流程
本发明属于药物制剂领域,具体涉及一种阿哌沙班药物组合物及其制备方法。
背景技术:
阿哌沙班(apixaban,商品名:艾乐妥、艾乐通/eliquis)是由百时美施贵宝和辉瑞联合开发的口服抗凝血药,是一种新型口服xa因子抑制剂,用于治疗包括深静脉血栓(deepvenousthrombosis,dvt)和肺栓塞(pulmonaryembolism,pe)在内的静脉血栓疾病。2011年5月,欧盟批准口服xa因子直接抑制剂阿哌沙班(商品名eliquis)上市,用于择期髋关节或膝关节置换手术的成年患者,以预防静脉血栓形成(venousthrombembolicevents,vte)。
阿哌沙班化学式为1-(4-甲氧基苯基)-7-氧代-6-[4-(2-氧代哌啶-1-基)苯基]-4,5,6,7-四氢-1h-吡唑并[3,4-c]吡啶-3-甲酰胺,分子式为c25h25n5o4,分子量为459.50,结构式如下:
阿哌沙班不溶于水,在水性介质(ph1.2-ph6.8)中溶解度为0.04mg/ml,存在溶解速度慢、体外溶出度低、生物利用度低的缺点,对药物的吸收有一定的影响。现有技术中已公开了多种方法用于提高阿哌沙班溶出度。
专利cn102770126a提供药物组合物中活性物质为n-1或h2-2的晶型,其包含具有等于或小于约89μm的d90(经激光散射方法测量)的晶状阿哌沙班颗粒以及药用稀释剂或载体,最优选d90小于25μm,采用干法制粒工艺制备。此发明是通过减小原料粒度增加溶出,但是粒度过小,往往会出现严重的团聚现象,不适合工业化生产。
专利cn102908324a将阿哌沙班制备成固体分散体,然后与处方量的微晶纤维素;交联聚维酮;微粉硅胶混合均匀直接压片。制备固体分散体如下:将聚乙二醇6000加热至60℃熔融,加入阿哌沙班剧烈搅拌为10000~20000r/min;对搅拌后的熔融物在-20℃的条件下迅速冷却0℃固化2h;固化物粉碎并过80目筛并20℃减压干燥4h,得到阿哌沙班固体分散体。此方法通过提高表面活性剂的用量来增加阿哌沙班的溶出度,使用了10-20份的表面活性剂聚乙二醇6000,大量的聚乙二醇给患者带来更大的副作用和不良反应。
因而寻求一种安全有效且适合工业化生产的方法来增加阿哌沙班的溶出度迫在眉睫。
技术实现要素:
本发明的目的在于提供一种阿哌沙班药物组合物,所得阿哌沙班制剂不仅稳定好、脆碎度小、溶出度高、体内生物利用度高、批次间溶出差异小、表面活性剂用量少,而且在制备过程也无团聚、粘冲现象适合工业化生产。
具体而言,本发明是通过如下技术方案实现的:
一种阿哌沙班药物组合物,包含乳糖、微晶纤维素、崩解剂、润滑剂,其中乳糖与微晶纤维素的重量比例为3~6:1,优选乳糖与微晶纤维素的重量比例为4~5:1。
在本发明的一个实施方案中,所述阿哌沙班的粒度d(0.9)≤30μm,优选d(0.9)≤25μm。
在本发明的一个实施方案中,所述的崩解剂选自交联羧甲纤维素钠、羧甲淀粉钠、低取代羟丙纤维素、交联聚维酮中的一种或多种,优选交联羧甲纤维素钠。
在本发明的一个实施方案中,所述的润滑剂选自硬脂酸镁、硬脂富马酸钠、滑石粉,优选硬脂酸镁。
在本发明的一个实施方案中,所述阿哌沙班药物组合物不含表面活性剂。
在本发明的一个实施方案中,所述阿哌沙班药物组合物,含有低于组合物总重量0.5%的表面活性剂,其中所述的表面活性剂是十二烷基硫酸钠、聚山梨酯、十二烷基苯磺酸钠中的一种或多种,优选十二烷基硫酸钠。
在本发明的一个实施方案中,包衣采用的包衣材料为薄膜包衣预混剂(胃溶型),例如
在本发明的一个实施方案中,所述阿哌沙班重量占组合物总重量的1%~10%,优选占组合物总重量的2.0%~7.5%。
在本发明的一个实施方案中,所述阿哌沙班重量占组合物总重量的3.0%。
在本发明的一个实施方案中,所述阿哌沙班重量占组合物总重量的6.0%。
在本发明的一个实施方案中,所述乳糖与微晶纤维素的重量占组合物总重量的60%~95%,优选占组合物总重量的80%~92%。
在本发明的一个实施方案中,所述的崩解剂其总重量占组合物总重量的2%~8%,优选占组合物总重量的3.0%~4.0%。
在本发明的一个实施方案中,所述的润滑剂,其占组合物总重量的0.1~2%,优选占组合物总重量的0.2~0.8%。
在本发明的一个实施方案中,所述的包衣增重占组合物总重量1%~6%,优选占组合物总重量2%~5%。
本发明的另一目的在于提供一种阿哌沙班药物组合物的制备方法,包括如下步骤:
(1)将阿哌沙班进行粉碎,控制其粒度d(0.9)≤30μm;
(2)在料斗混合机中,将乳糖、微晶纤维素、崩解剂与粉碎后与阿哌沙班进行混合,之后再与润滑剂进行混合;
(3)将步骤(2)所得中间体物料进行干法制粒,并与润滑剂混合。
(4)将步骤(3)所得混合物料加入到压片机中压片,控制素片硬度为3.00~10.00kg/cm2
(5)包衣。
在本发明的一个实施方案中,所述粉碎处理可以是用粉碎机进行粉碎。通常来说,可通过万能粉碎机、微粉碎机、涡轮粉碎机、球磨机或气流粉碎机等设备对原料进行粉碎,优选地采用万能粉碎机粉碎。
在本发明的一个实施方案中,先将步骤(2)中的乳糖及微晶纤维素过50-80目筛进行预处理,优选过60目~70目筛。
在本发明的一个实施方案中,当使用表面活性剂时,先将步骤(2)中的表面活性剂以粉碎机60~100目筛粉碎,优选过60目~80目筛。
在本发明的一个实施方案中,步骤(2)中,料斗混合机的转速为8rpm~20rpm,优选13rpm~18rpm,
在本发明的一个实施方案中,步骤(2)中,辅料与原料的混合时间为5min~30min,优选10min~25min。
在本发明的一个实施方案中,步骤(3)中,干法制粒机的制粒筛网孔径为0.8~1.5mm,优选0.8~1.0mm。
在本发明的一个实施方案中,步骤(4)中的压片机压片速度为10.0~100.0千片/小时,优选20.0~50.0千片/小时(压片开始和结束速度较低,中间多在40~50千片/小时)。
在本发明的一个实施方案中,步骤(4)中,压片机的主压力平均值为3.00~25.00kn优选7.00~11.00kn。
在本发明的一个实施方案中,步骤(4)中,所得素片硬度为4.00~7.00kg/cm2。
在本发明的一个实施方案中,具体包括如下步骤:
(1)将阿哌沙班进行粉碎,控制其粒度d(0.9)≤25μm;
(2)先将乳糖及微晶纤维素过60-70目筛进行预处理,在料斗混合机中,将乳糖、微晶纤维素、交联羧甲纤维素钠与粉碎后的阿哌沙班进行混合15min~25min,之后再将上述中间体物料与硬脂酸镁混合5min~15min。
(3)将步骤(2)所得中间体物料加入到干法制粒机中进行制粒,控制送料变频为8~30hz,压片变频为10~30hz,制粒变频为10~30hz。
(4)将步骤(3)所得中间体物料与硬脂酸镁混合,混合5min~15min。
(5)将步骤(4)所得的中间体物料加入到压片机中压片,控制压片速度为30.0~50.0千片/小时,主压力平均值为8.00~10.00kn,素片硬度为4.00~7.00kg/cm2。
(6)将素片加入包衣锅中进行包衣,控制片床温度为35~45℃,包衣增重至2~4%停止。
本发明所述阿哌沙班药物组合物的制备方法,颗粒流动性好,无团聚现象发生,且压片过程顺利,无粘冲;所得阿哌沙班片剂表面完整光洁、色泽均匀,无断裂、龟裂及粉碎现象。
附图说明
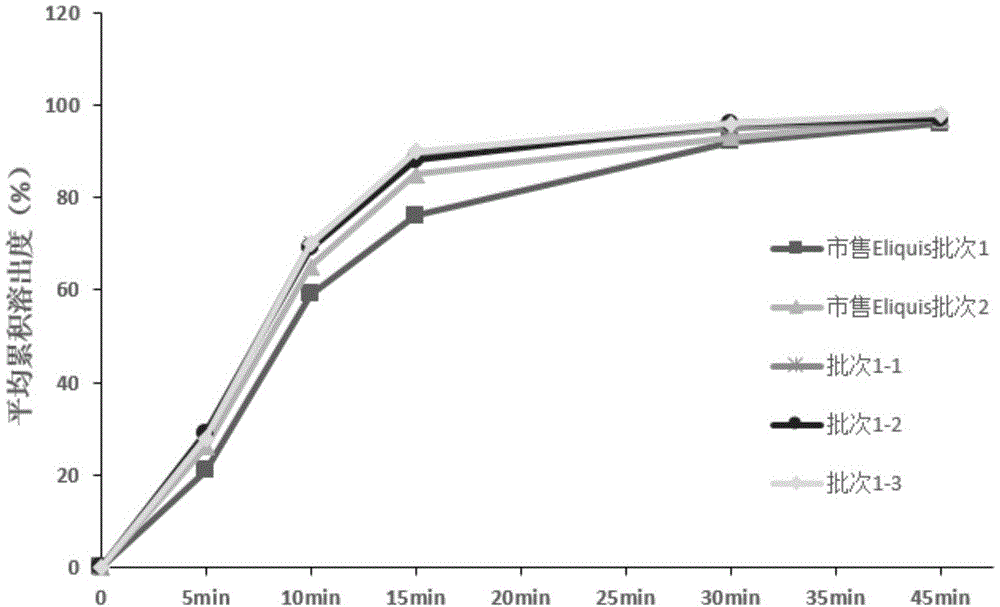
图1:实施例1的3批次片剂与市售eliquis在0.1mol/l盐酸溶液中的溶出行为。
图2:实施例1的3批次片剂与市售eliquis在ph6.8磷酸盐缓冲液中的溶出行为。
图3:实施例1的3批次片剂与市售eliquis在水中的溶出行为。
具体实施方式
为了进一步说明本发明,下面将结合具体实施例对本发明进行具体阐述,但本发明的保护范围并非限定于具体实施例。
实施例1
片剂处方:
制备工艺:
(1)原辅料预处理:将阿哌沙班粉碎,其d(0.9)为21.0μm,乳糖及微晶纤维素过60目筛处理,十二烷基硫酸钠60目筛粉碎;
(2)将处方量的无水乳糖、微晶纤维素、十二烷基硫酸钠、交联羧甲纤维素钠以及阿哌沙班加入至固定料斗混合机中,设定转速10rpm,混合20min
(3)将(2)中所得中间体物料与内加硬脂酸镁混合5min。
(4)将(3)中所得中间体物料加入至干法制粒机中制粒,送料变频在8~30hz范围内调整,压片变频在10~30hz范围内调整,制粒变频在10~30hz范围内调整,整粒筛网为1.0mm。
(5)将(4)中所得中间体物料加入至料斗混合机中,并将处方量的硬脂酸镁加入至料斗混合机中,设定转速10rpm,混合15min;
(6)压片,设定旋转压片机的压片速度为30.0千片/小时,填料器转速90rpm,主压力平均值7.9~8.5kn,所得素片硬度为5.45kg/cm2。
(7)包衣:将素片加入包衣锅中,控制包衣温度为35~45℃,待包衣增重至3.0%后停止包衣。
实施例2
片剂处方:
制备工艺:
(1)原辅料预处理:将阿哌沙班粉碎,其d(0.9)为20.1μm,乳糖及微晶纤维素过60目筛处理,十二烷基硫酸钠60目筛粉碎;
(2)将处方量的无水乳糖、微晶纤维素、十二烷基硫酸钠、交联羧甲纤维素钠以及阿哌沙班加入至固定料斗混合机中,设定转速10rpm,混合20min
(3)将(2)中所得中间体物料与内加硬脂酸镁混合5min。
(4)将(3)中所得中间体物料加入至干法制粒机中制粒,送料变频在8~30hz范围内调整,压片变频在10~30hz范围内调整,制粒变频在10~30hz范围内调整,整粒筛网为1.0mm。
(5)将(4)中所得中间体物料加入至料斗混合机中,并将处方量的硬脂酸镁加入至料斗混合机中,设定转速10rpm,混合15min;
(6)压片,设定旋转压片机的压片速度为30.0千片/小时,填料器转速90rpm,主压力平均值7.9~8.5kn,所得素片硬度为5.75kg/cm2。
(7)包衣:将素片加入包衣锅中,控制包衣温度为35~45℃,待包衣增重至3.0%后停止包衣。
实施例3
片剂处方:
制备工艺:
(1)原辅料预处理:将阿哌沙班粉碎,其d(0.9)为12.8μm,乳糖及微晶纤维素过70目筛处理,十二烷基硫酸钠80目筛粉碎;
((2)将处方量的无水乳糖、微晶纤维素、交联羧甲纤维素钠以及阿哌沙班加入至固定料斗混合机中,设定转速10rpm,混合20min
(3)将(2)中所得中间体物料与内加硬脂酸镁混合5min。
(4)将(3)中所得中间体物料加入至干法制粒机中制粒,送料变频在8~30hz范围内调整,压片变频在10~30hz范围内调整,制粒变频在10~30hz范围内调整,整粒筛网为1.0mm。
(5)将(4)中所得中间体物料加入至料斗混合机中,并将处方量的硬脂酸镁加入至料斗混合机中,设定转速10rpm,混合15min;
(6)压片,设定旋转压片机的压片速度为50.0千片/小时,填料器转速80rpm,主压力平均值10.6~11.5kn,所得素片硬度为6.90kg/cm2。
(7)包衣:将素片加入包衣锅中,控制包衣温度为35~45℃,待包衣增重至2.5%后停止包衣。
实施例4
片剂处方:
制备工艺:
(1)原辅料预处理:将阿哌沙班微粉,其d(0.9)为25.5μm,乳糖及微晶纤维素过70目筛处理;
(2)将处方量的无水乳糖、微晶纤维素、交联羧甲纤维素钠以及阿哌沙班加入至固定料斗混合机中,设定转速10rpm,混合20min
(3)将(2)中所得中间体物料与内加硬脂酸镁混合5min。
(4)将(3)中所得中间体物料加入至干法制粒机中制粒,送料变频在8~30hz范围内调整,压片变频在10~30hz范围内调整,制粒变频在10~30hz范围内调整,整粒筛网为1.0mm。
(5)将(4)中所得中间体物料加入至料斗混合机中,并将处方量的硬脂酸镁加入至料斗混合机中,设定转速10rpm,混合15min;
(6)压片,设定旋转压片机的压片速度为40.0千片/小时,填料器转速80rpm,主压力平均值7.2~9.0kn,所得素片硬度为5.81kg/cm2。
(7)包衣:将素片加入包衣锅中,控制包衣温度为35~45℃,待包衣增重至4.0%后停止包衣。
实施例5
片剂处方:
制备工艺:
(1)原辅料预处理:将阿哌沙班微粉,其d(0.9)为18.4μm,乳糖及微晶纤维素过60目筛处理;
(2)将处方量的无水乳糖、微晶纤维素、羧甲淀粉钠以及阿哌沙班加入至固定料斗混合机中,设定转速10rpm,混合20min
(3)将(2)中所得中间体物料与内加硬脂酸镁混合5min。
(4)将(3)中所得中间体物料加入至干法制粒机中制粒,送料变频在8~30hz范围内调整,压片变频在10~30hz范围内调整,制粒变频在10~30hz范围内调整,整粒筛网为1.0mm。
(5)将(4)中所得中间体物料加入至料斗混合机中,并将处方量的硬脂酸镁加入至料斗混合机中,设定转速10rpm,混合15min;
(6)压片,设定旋转压片机的压片速度为30.0千片/小时,填料器转速90rpm,主压力平均值7.9~8.5kn,所得素片硬度为5.65kg/cm2。
(7)包衣:将素片加入包衣锅中,控制包衣温度为35~45℃,待包衣增重至3.0%后停止包衣。
实施例5脆碎度考察
根据2015年版中国药典四部“0923片剂脆碎度检查法”的方法,测定实施例1-4所制得的素片的脆碎度。
表1
结果显示,全部批次的片剂均未出现断裂、龟裂及粉碎的情形,并且平均减失重量均<0.3%,表明这些片剂具有优良的脆碎度。
实施例6药物溶出度考察
溶出介质:0.1mol/l盐酸、ph6.8磷酸盐缓冲溶液以及水,900ml;
溶出方法:桨法,50rpm;
介质温度:37.0℃±0.5℃;
取样:分别经5min、10min、15min、30min、45min时,取样10ml,经聚醚砜膜过滤,同时补加等温同种介质;
样品:从实施例1-5中随机抽取5个样品。
测定结果如表2。
表2
实施例7稳定性考察
随机抽取实施例1-4的样品,将其在加速40℃±2℃/rh75%±5%条件下放置6个月,观察有关物质的变化,具体结果如表3。
表3
实施例8批次间溶出度考察
完全按照实施例1的处方及工艺,连续生产3批制剂样品,对应为批次1-1、批次1-2、批次1-3,从每批中随机抽取30片作为样品,考察每批样品在不同溶出介质中的溶出情况。
测定结果如附图1-3所示。
由各批次溶出曲线对比可以看出,市售eliquis其15min前的溶出波动较大,而本发明实施例1各批次溶出行为几乎相同,差异性小,适宜工业化大生产。
实施例2-5批次间的溶出与实施例1的类似。
- 还没有人留言评论。精彩留言会获得点赞!