3,5-二氨基-6-氯-N-(N-(4-苯基丁基)甲脒基)吡嗪-2-甲酰胺化合物的制作方法
本发明涉及可用作钠通道阻断剂的新的经取代的3,5-二氨基-6-氯-N-(N-(4-芳基丁基)甲脒基)吡嗪-2-甲酰胺化合物,特别地包括经取代的3,5-二氨基-6-氯-N-(N-(4-苯基丁基)甲脒基)吡嗪-2-甲酰胺化合物,例如,3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺及其对映体和可药用盐,包含所述化合物的组合物,所述化合物的治疗方法和用途以及用于制备所述化合物的方法。
背景技术:
在环境与身体之间交界面的黏膜表面进化出了多种“先天防御”,即保护机制。这些先天防御的主要形式是用液体清洁这些表面。通常来说,黏膜表面上液体层的量反映上皮液体分泌与上皮液体吸收之间的平衡,上皮液体分泌通常反映与水(和阳离子反离子(cation counter-ion))偶联的阴离子(Cl-和/或HCO3-)分泌,上皮液体吸收通常反映与水和反阴离子(Cl-和/或HCO3-)偶联的Na+吸收。黏膜表面的许多疾病是由这些黏膜表面上保护性液体太少造成的,而后者由分泌(太少)与吸收(相对太多)之间失衡造成。表征这些黏膜功能障碍的缺陷的盐运输过程存在于黏膜表面的上皮层中。
补充黏膜表面上的保护性液体层的一种方法是通过阻断Na+通道和液体吸收来使系统“再平衡”。介导Na+和液体吸收的限速步骤的上皮蛋白质是上皮Na+通道(“ENaC”)。ENaC位于上皮的顶面上,即黏膜表面-环境交界面。理想的是,为了抑制ENaC介导的Na+和液体吸收,必须将阿米洛利(amiloride)类ENaC阻断剂递送至黏膜表面,并且保持在该部位以实现最大治疗益处。
已经报道ENaC阻断剂用于通过提高黏膜水化来改善的多种疾病。特别地,已经报道了ENaC阻断剂用于治疗呼吸疾病,例如囊性纤维化(cystic fibrosis,CF)和COPD,包括慢性支气管炎(chronic bronchitis,CB)和肺气肿,所述疾病反映为机体不能正常地从肺部清除黏液,最终导致慢性气道感染。参见Evidence for airway surface dehydration as the initiating event in CF airway disease,R.C.Boucher,Journal of Internal Medicine,第261卷,第1期,2007年1月,第5-16页;和Cystic fibrosis:a disease of vulnerability to airway surface dehydration,R.C.Boucher,Trends in Molecular Medicine,第13卷,第6期,2007年6月,第231-240页。
数据表明CB和CF二者的初始问题都是无法清理气道表面的黏液。无法清理黏液反映了作为气道表面上的气道表面液体(airyway surface liquid,ASL)的黏液的量失衡。这种失衡造成ASL相对减少,这导致黏液浓缩、纤周液体(periciliary liquid,PCL)的润滑活性降低、黏液黏附于气道表面以及无法通过纤毛活动将黏液清除到口腔。黏液清除的降低导致黏附于气道表面的黏液的长期细菌定殖。细菌的长期滞留、在长期的基础上局部抗微生物物质无法杀死陷入黏液的细菌,以及随后对于这种类型的表面感染的慢性炎性应答显示为CB和CF。
对于特异性治疗通过提高黏膜水化来缓解的多种疾病(包括CB、COPD和CF等)的产品,目前有巨大的未满足的医疗需要。目前对于CB、COPD和CF的治疗集中于治疗这些疾病的症状和/或晚期影响。但是,这些治疗中没有一种有效地解决无法从肺部清除黏液的根本问题。
R.C.Boucher在U.S.6,264,975中描述了使用吡嗪酰胍(pyrazinoylguanidine)钠通道阻断剂使黏膜表面水化,以公知的利尿剂阿米洛利、苯扎米尔(benzamil)和非那米尔(phenamil)为代表。然而,考虑到可吸入肺的药物的重量有限;(2)迅速吸收,以及由此表现为在黏膜表面上的不期望的短的半衰期;以及(3)从ENaC自由解离,这些化合物相对弱效。需要在黏膜表面上具有较长半衰期的更有效药物。
在其他黏膜表面上的保护性表面液体太少是多种疾病的通常病理生理学。例如,在口干燥症(口干)中,口腔缺少液体是因为腮腺、舌下腺和颌下腺无法分泌液体,而持续的Na+(ENaC)运输介导从口腔吸收液体。干性角膜结膜炎(keratoconjunctivitis sira)(干眼)由在结膜表面上连续Na+依赖的液体吸收的情况下泪腺无法分泌液体导致。在鼻窦炎中,具有黏蛋白分泌与相对ASL消耗之间的失衡。在近端的小肠中无法分泌Cl-(和液体)与在末端回肠中Na+(和液体)的吸收提高一起,导致远端肠梗阻综合征(distal intestinal obstruction syndrome,DIOS)。在老年患者中,在降结肠中过量的Na+(和体积)吸收产生便秘和憩室炎(diverticulitis)。
公开的文献包括针对吡嗪酰胍类似物作为钠通道阻滞剂的若干专利申请和授权专利。这些出版物的实例包括PCT公开No.WO2003/070182、WO2003/070184、WO2004/073629、WO2005/025496、WO2005/016879、WO2005/018644、WO2006/022935、WO2006/023573、WO2006/023617、WO2007/018640、WO2007/146869、WO2008/031028、WO2008/031048、以及美国专利No.6858614、6858615、6903105、6,995,160、7,026,325、7,030,117、7064129、7186833、7189719、7192958、7192959、7192960、7241766、7247636、7247637、7317013、7332496、7,345,044、7368447、7368450、7368451、7375107、7388013、7399766、7410968、7,745,442、7807834、7,820,678、7842697、7868010、7,875,619、7,956,059、7,981,898、8,008,494、8022,210、8,058,278、8,124,607、8,143,256、8,163,758、8,198,286、和8,211,895。
依然需要在黏膜组织上具有增强的效力和有效性的新的钠通道阻断化合物。依然还需要提供治疗作用但是使接受者中高钾血症之发生或发展尽可能小或使其消除的新的钠通道阻断化合物。
发明概述
本发明提供了式(A)的化合物或其可药用盐:
其中Ar是选自以下的部分:
X选自-CH2-、-O-或-S-;
R1和R2独立地选自H和C1-C6烷基;
或者R1和R2与其所结合的氮原子一起形成5元或6元杂环,所述杂环任选地包含一个另外的选自N或O的环杂原子;
R3是具有3至8个碳原子的烷基或者具有3至8个碳原子的多羟基化烷基;
R4是具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
本发明还提供了3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的溶剂合物和水合物,单独的立体异构体,包括光学异构体(对映体和非对映体)和几何异构体(顺式/反式异构),立体异构体的混合物以及互变异构体,或其可药用盐,以及包含所述化合物或其可药用盐的药物组合物,其在治疗方法中的用途及其制备的方法。
附图简述
通过参考结合以下附图的本文中的信息,可更容易地获得对本发明及其许多优点的更全面的理解:
图1是证明与载剂相比化合物II-d的4小时剂量响应的曲线图。
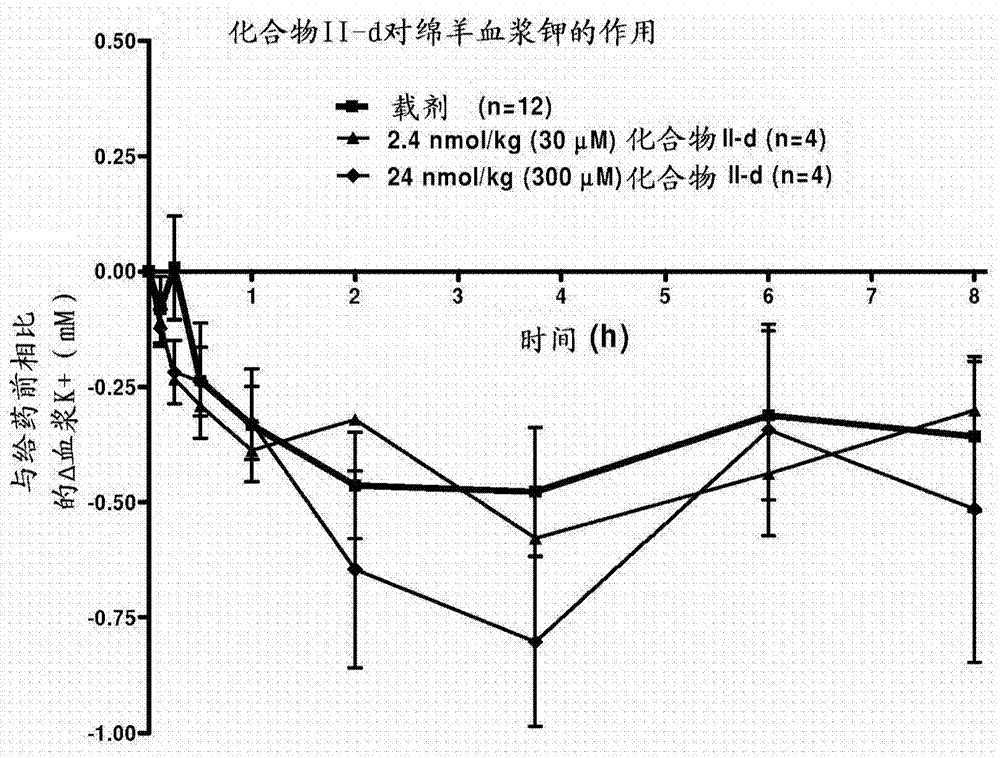
图2是化合物II-d对绵羊血浆的作用的曲线图。
图3是证明给药后4小时化合物II-d对绵羊MCC之作用的曲线图。
图4是证明给药后4小时比较实施例1对绵羊MCC之作用的曲线图。
图5是证明比较实施例1对绵羊血浆钾之作用的曲线图。
图6是证明给药后4小时化合物II-d和比较实施例1对绵羊MCC之作用的曲线图。
图7是证明化合物II-d和比较实施例1对绵羊血浆钾之作用的曲线图。
发明详述
还提供了包括由式(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)独立地表示的12组化合物或其可药用盐的实施方案:
其中,在每个组(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)中:
R1和R2独立地选自H和C1-C6烷基;
或者R1和R2与其所结合的氮原子一起形成5元或6元杂环,所述杂环任选地包含一个另外的选自N或O的环杂原子;R3是具有3至8个碳原子的烷基或具有3至8个碳原子的多羟基化烷基;
R4是具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2独立地选自H和C1-C6烷基;
R3和R4各自独立地为具有3至8个碳原子的多羟基化烷基;
R5选自H或C1-C3烷基。
在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2独立地选自H和C1-C6烷基;
R3是具有3至8个碳原子的烷基;并且
R4是具有3至8个碳原子的多羟基化烷基;
R5选自H或C1-C3烷基。
此外,在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2独立地选自H和-CH3;
R3是具有3至8个碳原子的烷基或具有3至8个碳原子的多羟基化烷基;
R4是具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2独立地选自H和-CH3;
R3是具有3至8个碳原子的烷基;
R4是具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
再者,在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2独立地选自H和-CH3;
R3和R4各自独立地为具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
包括在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2为H;
R3是具有3至8个碳原子的烷基或具有3至8个碳原子的多羟基化烷基;
R4是具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
此外,在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中:
R1和R2为H;
R3是具有3至8个碳原子的烷基;
R4是具有3至8个碳原子的多羟基化烷基;并且
R5选自H或C1-C3烷基。
仍进一步在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)表示的每个化合物组中,存在另外的化合物组或其可药用盐,其中R1和R2为H;并且R3和R4各自独立地为具有3至8个碳原子的多羟基化烷基;并且R5选自H或C1-C3烷基。
在上文中所述的每个组中,存在其中R5为H的另外的组;或其可药用盐。在上文中所述的每个组中,还存在其中R5为-CH3的另外的组;或其可药用盐。
由R1和R2与其所结合的氮原子一起形成的任选地包含一个另外的选自N或O的环杂原子的5元或6元杂环包括吡咯烷基环、哌啶基环、哌嗪基环和吗啉基环。
本发明的多羟基化烷基是其中3至8个碳原子被两个或更多个羟基取代的烷基链的那些。多羟基化烷基的实例是丁烷-1,4-二醇、丁烷-1,2,2-三醇、丁烷-1,1,2,3-四醇、戊烷-1,2,3,4-四醇、己烷-1,2,3,4,5-五醇、庚烷1,2,3,4,5,6-六醇、和辛烷1,2,3,4,5,6,7-七醇。
在本文中所述的每个化合物组中的一个实施方案是其中所述多羟基化烷基具有式-CH2-(CHR5)n-H的那些化合物,其中n是选自2、3、4、5、6或7的整数并且在每种情况下R5独立地为H或OH,前提是至少两个R5基团是OH。
在本文中所述的每个化合物组中的另一个实施方案是其中所述多羟基化烷基具有式-CH2-CHOH-(CHR6)m-H的那些化合物,其中m是选自1、2、3、4、5或6的整数并且在每种情况下R6独立地为H或OH,前提是至少一个R6基团是OH。
在本文中所述的每个化合物组中的又一个实施方案包括其中所述多羟基化烷基具有式-CH2-(CHOH)n-CH2OH的化合物,其中n是选自1、2、3、4、5或6的整数。在本文中所述的各化合物组中的另一个实施方案包括其中n是选自2、3、4或5的整数的化合物。在每个组中的另一个实施方案包括其中n是选自3、4或5的整数的化合物。
在本文中所述的每个化合物组中的另一个实施方案中,由R4式-CH2-(CHOH)n-CH2OH表示的链是2,3,4,5,6-五羟基己烷,具有下式:
在本文中所述的每个化合物组中的又一个实施方案中,由R4式-CH2-(CHOH)n-CH2OH表示的链是下式的链:
在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)的化合物独立地表示的每个组中,存在另外的实施方案或其可药用盐,其中:R1为H;R2为H或C1-C3烷基;
R3是具有4至8个碳原子的烷基或具有4至8个碳原子的多羟基化烷基;并且
R4是式-CH2-(CHOH)n-CH2OH的多羟基化烷基;并且
在每种情况下,n独立地为选自1、2、3、4、5或6的整数。
在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)的化合物独立地表示的每个组中,仍然存在另外的实施方案或其可药用盐,其中R1和R2为H;R3是具有5至7个碳原子的烷基;R4是式-CH2-(CHOH)n-CH2OH的多羟基化烷基;并且n是选自1、2、3、4、5或6的整数。
在本文中所述的每个组中,存在另外的实施方案,其中R4是式-CH2-(CHOH)n-CH2OH的多羟基化烷基并且n是选自3、4或5的整数。在每个组中的又一个实施方案中,R4是式-CH2-(CHOH)n-CH2OH的多羟基化烷基并且n是4。
在由式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)和(M)的化合物独立地表示的每个组中,仍然存在另外的实施方案或其可药用盐,其中:R1和R2为H;R3是具有6个碳原子的烷基;R4是式-CH2-(CHOH)n-CH2OH的多羟基化烷基;并且n是4。
还提供了式(B-1)的化合物3,5-二氨基-N-(N-(4-(4-(4-氨基-3-(3-(双(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐:
在另一个实施方案中,式(A)的化合物为具有式(B-2)的3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐:
还提供了式(B-3)的化合物3,5-二氨基-N-(N-(4-(4-(4-氨基-3-(3-(己基(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐:
在另一个实施方案中,化合物为具有式(B-4)的3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐:
本发明的另一些化合物包括式(E-1)、(E-2)、(E-3)和(E-4)的那些或其可药用盐:
3,5-二氨基-N-(N-(4-(6-(3-氨基-2-(3-(双(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;
3,5-二氨基-N-(N-(4-(6-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;
3,5-二氨基-N-(N-(4-(6-(4-氨基-3-(3-(己基(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;以及
3,5-二氨基-N-(N-(4-(6-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺。
本发明的另一些化合物包括式(H-1)、(H-2)、(H-3)和(H-4)的那些或其可药用盐:
3,5-二氨基-N-(N-(4-(4-((3R)-4-氨基-3-(3-(双(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;以及
3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;以及
3,5-二氨基-N-(N-(4-(4-((3R)-4-氨基-3-(3-(己基(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;以及
3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺。
本发明的另一些化合物包括式(K-1)、(K-2)、(K-3)和(K-4)的那些或其可药用盐:
3,5-二氨基-N-(N-(4-(4-(4-氨基-3-(3-(双(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;
3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;
3,5-二氨基-N-(N-(4-(4-(3-氨基-2-(3-(己基(2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺;以及
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺。
如本文所使用的,以下术语为所指定的定义。
“本发明的化合物”意指式(A)化合物或其盐,特别是其可药用盐。
“式(A)化合物”意指具有本文指定为式(A)之结构式的化合物。式(A)化合物包括溶剂合物和水合物(即,式(A)化合物与溶剂的加合物)。在其中式(A)化合物包含一个或更多个手性中心的那些实施方案中,该用语旨在涵盖每个单独的立体异构体,包括光学异构体(对映体和非对映体)和几何异构体(顺式/反式异构)以及立体异构体的混合物。此外,式(A)化合物还包括所示式的互变异构体。
在整个说明书和实施例中,使用标准IUPAC命名原则命名化合物,其中可能包括使用CambridgeSoft Corp./PerkinElmer所销售的ChemDraw Ultra 11.0软件程序对化合物进行命名。
在一些化学结构表示中,未描绘出碳原子具有足够的连接变量以得到四价时,则假定需要提供四价的其余碳取代基为氢。类似地,在一些化学结构中,画出了键而未指出末端基团,根据本领域中的惯例,这样的键表示甲基(Me、-CH3)。
式I化合物可以是游离碱或盐(特别是可药用盐)的形式。可药用盐的综述参见Berge等,J.Pharma Sci.(1977)66:1-19。
由无机酸或有机酸形成的可药用盐包括例如:盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、硫酸氢盐、硝酸盐、氨基磺酸盐、磷酸盐、磷酸氢盐、乙酸盐、三氟乙酸盐、马来酸盐、苹果酸盐、富马酸盐、乳酸盐、酒石酸盐、柠檬酸盐、甲酸盐、葡糖酸盐、琥珀酸盐、丙酮酸盐、鞣酸盐、抗坏血酸盐、棕榈酸盐、水杨酸盐、硬脂酸盐、邻苯二甲酸盐、藻酸盐、聚谷氨酸盐、草酸盐、草酰乙酸盐、糖二酸盐、苯甲酸盐、烷基或芳基磺酸盐(例如,甲磺酸盐、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐或萘磺酸盐)和异硫代硫酸盐(isothionate);与氨基酸形成的复合物,所述氨基酸例如赖氨酸、精氨酸、谷氨酸、甘氨酸、丝氨酸、苏氨酸、丙氨酸、异亮氨酸、亮氨酸等。本发明的化合物还可以是由元素的阴离子例如氯离子、溴离子或碘离子所形成的盐的形式。
为了治疗用途,式(A)化合物的活性成分的盐是可药用的,即,其是来源于可药用酸的盐。然而,也可见到不可药用酸的盐用于例如可药用化合物的制备或纯化中。例如,三氟乙酸可用于这样的应用。所有盐,无论是否来源于可药用酸,均在本发明的范围内。
术语“手性”是指具有与镜像体(mirror image partner)不重叠特性的分子,而术语“非手性”是指可与其镜像体重叠的分子。
术语“立体异构体”是指具有相同化学组成但原子或基团在空间排列上不同的化合物。“非对映体”是指具有两个或更多个手性中心并且其分子不是彼此镜像的立体异构体。非对映体具有不同的物理性质,例如熔点、沸点、谱性质和反应性。可以在高分辨率分析操作(例如电泳法和色谱法)下分离非对映体的混合物。“对映体”是指与彼此的镜像不重叠的化合物的两种立体异构体。
本文中使用的立体化学的定义和规则通常遵循S.P.Parker,Ed.,McGraw-Hill Dictionary of Chemical Terms(1984)McGraw-Hill Book Company,New York;以及Eliel,E.和Wilen,S.,Stereochemistry of Organic Compounds(1994)John Wiley&Sons,Inc.,New York。
在本文结构中使用的波状或波形符号理解为表示通过它将所示结构键合到分子的另一部分的点。
许多有机化合物以光学活性形式存在,即,它们具有使平面偏振光的平面旋转的能力。在对光学活性化合物的描述中,使用前缀D和L或者R和S表示分子关于其手性中心的绝对构型。特定立体异构体还可以称作对映体,这样的异构体的混合物通常称作对映体混合物。对映体的50∶50混合物被称为外消旋混合物或外消旋体,其可以在化学反应或过程中没有立体选择性或立体特异性时发生。术语“外消旋混合物”或“外消旋体”是指两种对映体物质的等摩尔混合物。
术语“互变异构体”是指其中氢原子的迁移导致两种或更多种结构的立体异构体类型。式(A)化合物可以以不同的互变异构形式存在。本领域技术人员应承认,脒、酰胺、胍、脲、硫脲、杂环等可以以互变异构形式存在。作为举例而不是限制,式(A)化合物可以以如下所示的多种互变异构形式存在:
式(A)的所有实施方案之脒、酰胺、胍、脲、硫脲、杂环等的所有互变异构形式均在本发明的范围内。互变异构体以平衡状态存在,因此本领域技术人员应理解,对所提供的式中单个互变异构体的描述等于是指所有可能的互变异构体。
应注意的是,式(A)范围内的化合物的所有对映体、非对映体和外消旋体混合物、互变异构体、多晶型物、假多晶型物及其可药用盐均涵盖在本发明中。这些对映体和非对映体的所有混合物,包括对映体富集的混合物和非对映体富集的混合物,均在本发明的范围内。对映体富集的混合物是其中指定对映体与备选对映体的比率大于50∶50之对映体的混合物。更特别地,对映体富集的混合物包含至少约75%的指定对映体,并且优选地至少约85%的指定对映体。在一个实施方案中,对映体富集的混合物基本上不含其他对映体。类似地,非对映体富集的混合物是其中指定非对映体的量大于每个备选非对映体的量之非对映体的混合物。更特别地,非对映体富集的混合物包含至少约75%的指定非对映体,并且优选地至少约85%的指定非对映体。在一个实施方案中,非对映体富集的混合物基本上不含所有其他非对映体。本领域技术人员将理解,术语“基本上不含”表示存在少于5%的其他非对映体,优选地少于1%,更优选地少于0.1%。在另一些实施方案中,不存在其他非对映体或者任何其他非对映体的存在量低于检测水平。可通过本领域已知的技术包括高效液相色谱法(HPLC)和手性盐的结晶来分离立体异构体。
可通过使用例如用光学活性拆分剂形成非对映体的方法拆分外消旋混合物来获得单一立体异构体(例如基本上不含其立体异构体的对映体)(″Stereochemistry of Carbon Compounds,″(1962)by E.L.Eliel,McGraw Hill;Lochmuller,C.H.,(1975)J.Chromatogr.,113:(3)283-302)。可以通过任何合适的方法使本发明手性化合物的外消旋混合物分开和分离,包括:(1)与手性化合物形成离子非对映体盐,并通过分级结晶或其他方法分离,(2)与手性衍生试剂形成非对映体化合物,分离非对映体并转化成纯的立体异构体,以及(3)在手性条件下直接分离基本纯的或富集的立体异构体。
在一个实施方案中,本发明提供了作为主要异构体的具有式(B-2)的3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐的对映体富集的混合物,或者包含其对映体富集的混合物的组合物。
另一个实施方案提供了作为主要异构体的具有式(B-4)的3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐之对映体富集的混合物,或包含其对映体富集的混合物的组合物。
另一个实施方案提供了作为主要异构体的具有式(H-2)的3,5-二氨基-N-(N-(4-(4-((R)-4-氨基-3-(3-(双((2R,3S,4S,5S)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐的对映体富集的混合物,或包含其对映体富集的混合物的组合物。
又一个实施方案提供了作为主要异构体的具有式(H-4)的3,5-二氨基-N-(N-(4-(4-((R)-4-氨基-3-(3-(己基((2R,3S,4S,5S)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺或其可药用盐的对映体富集的混合物,或包含其对映体富集的混合物的组合物。
另一些实施方案包括对映体富集的混合物或组合物,其分别包含式(B-2)、(B-4)、(H-2)和(H-4)的化合物或其可药用盐作为其各自混合物的每一种中的主要异构体。
另一些实施方案包括对映体富集的混合物或组合物,其分别包含式(B-2)、(B-4)、(H-2)和(H-4)的化合物或其可药用盐,基本不含其各自混合物的每一种中的其他异构体。
式(A)化合物及其可药用盐可作为不同的多晶型物或假多晶型物存在。如本文所使用的,结晶性多晶型物意指结晶化合物以不同晶体结构存在的能力。结晶性多晶型物可由结晶堆积(堆积多晶型)中的差异或相同分子不同构象异构体(conformer)之间的堆积差异(构象多晶型)造成。如本文中使用的,结晶性假多晶型物也包括化合物的水合物或溶剂合物以不同晶体结构存在的能力。本发明的假多晶型物可因为结晶堆积中的差异(堆积假多晶型)或相同分子不同构象异构体之间的堆积差异(构象假多晶型)而存在。本发明包括式(A)化合物及其可药用盐的所有多晶型物和假多晶型物。
式(A)化合物及其可药用盐还可以作为无定形固体存在。如本文中使用的,无定形固体是在固体中没有长程有序(long-range order)的原子位置的固体。当晶体大小为2纳米或更小时,该定义同样适用。可使用包含溶剂的添加剂以产生本发明的无定形形式。包括本文中所述的所有药物组合物、治疗方法、组合产品及其用途的本发明包括式(A)化合物及其可药用盐的所有无定形形式。
用途
本发明化合物表现出作为钠通道阻断剂的活性。不限于任何特定的理论,认为本发明化合物可在体内通过阻断存在于黏膜表面中上皮钠通道从而降低黏膜表面对水的吸收来起作用。该作用增多了黏膜表面上保护性液体的体积并使系统重新平衡。
因此,本发明的化合物可用作药物,特别是用于治疗适用于钠通道阻断剂的临床病症。这样的病症包括在有此需要的人中的肺部病症,例如与可逆或不可逆的气道阻塞相关的疾病、慢性阻塞性肺病(COPD)(包括COPD的急性加剧)、哮喘、支气管扩张(包括因囊性纤维化以外的病症引起的支气管扩张)、急性支气管炎、慢性支气管炎、病毒感染后的咳嗽(post-viral cough)、囊性纤维化、肺气肿、肺炎、全细支气管炎、移植相关细支气管炎(包括肺移植和骨髓移植相关支气管炎)。本发明化合物还可用于在使用通气机的患者(ventilated patient)中治疗通气机相关气管支气管炎和/或预防通气机相关肺炎。本发明包括用于在有此需要的哺乳动物中(优选在有此需要的人中)治疗这些病症中每一种的方法,每一种方法包括向所述哺乳动物施用药用有效量的本发明化合物或其可药用盐。还提供了(a)用于在有此需要的哺乳动物中减缓COPD恶化的方法;(b)用于在有此需要的哺乳动物中减缓CF恶化的方法;(c)在有此需要的哺乳动物中改善肺功能(FEV1)的方法;(d)在患有COPD的哺乳动物中改善肺功能(FEV1)的方法;(e)在患有CF的哺乳动物中改善肺功能(FEV1)的方法;(f)在有此需要的哺乳动物中减轻气道感染的方法。
还提供了在哺乳动物中刺激、增强或改善黏膜纤毛清除的方法,所述方法包括向有此需要的哺乳动物施用药用有效量的式(A)化合物或其可药用盐。黏膜纤毛清除应被理解为包括参与在气道中转移或清除黏液的天然黏膜纤毛作用,包括支气管的自清除(self-clearing)机制。因此,还提供了在有此需要的哺乳动物的气道中改善黏液清除的方法。
此外,钠通道阻断剂可适用于治疗通过提高除肺黏膜表面以外之黏膜表面的黏膜水化来改善的病症。这样的病症的实例包括干口(口干燥症)、皮肤干燥、阴道干燥、窦炎、鼻窦炎、鼻脱水(包括因施用干燥氧所致鼻脱水)、干眼、舍格伦病(Sjogren’s disease)、中耳炎、原发性纤毛运动障碍(primary ciliary dyskinesia)、远端肠梗阻综合征、食管炎、便秘和慢性憩室炎。本发明化合物还可用于促进眼或角膜水化。
本发明化合物还可用于从人获得痰样品的方法。所述方法可如下进行:向患者的至少一个肺施用本发明的化合物,然后诱导和收集来自该人的痰样品。
因此,在一个方面,本发明提供了用于在哺乳动物(例如人)中治疗适用于钠通道阻断剂之病症的方法。
在另一些实施方案中,本发明提供的每一种本文中所述方法具有使所述方法的接受者中的高钾血症的尽可能小或被消除的额外益处。还提供了包括每一种本文中所述方法的一些实施方案,在其中实现了治疗指数改善。
如本文所使用的术语“治疗”是指逆转、缓解、抑制疾病或病症或者这些疾病或病症的一种或更多种症状的进展,或者预防它们。
本文描述的所有治疗性方法均通过向有治疗需要的对象(通常是哺乳动物,并且优选人)施用有效量的本发明化合物、式(A)化合物或其可药用盐。
在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗通过提高黏膜水化改善的病症的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗与可逆的或不可逆的气道阻塞相关的疾病的方法。在一个特定的实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗慢性阻塞性肺病(COPD)的方法。在一个特定的实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中降低COPD的急性加剧的频率、严重程度或持续时间或者用于治疗COPD的急性加剧的一种或更多种症状的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗哮喘的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗支气管扩张(包括囊性纤维化以外的病症引起的支气管扩张)的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗支气管炎(包括急性和慢性支气管炎)的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗病毒感染后的咳嗽的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗囊性纤维化的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗肺气肿的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗肺炎的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗全细支气管炎的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗移植相关细支气管炎(包括肺移植和骨髓移植相关细支气管炎)的方法。在一个实施方案中,本发明提供了用于在有此需要的使用通气机的人中治疗通气机相关气管支气管炎和/或预防通气机相关肺炎的方法。
本发明提供了用于在有此需要的人中治疗疾病或预防通气机相关肺炎的特定方法,所述疾病选自可逆或不可逆的气道阻塞、慢性阻塞性肺病(COPD)、哮喘、支气管扩张(包括囊性纤维化以外的病症引起的支气管扩张)、急性支气管炎、慢性支气管炎、病毒感染后的咳嗽、囊性纤维化、肺气肿、肺炎、全细支气管炎、移植相关细支气管炎和通气机相关气管支气管炎,每一种方法包括向所述人施用有效量的式(B-2)化合物或其可药用盐。在每一种治疗方法的另一些实施方案中,可药用盐形式是式(B-2)化合物的盐酸盐或羟基萘甲酸盐。在每一种治疗方法中的另一些实施方案中,使用式(B-2)化合物的游离碱。
在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗干口(口干燥症)的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗皮肤干燥的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗阴道干燥的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗窦炎、鼻窦炎或鼻脱水(包括因施用干燥氧所致鼻脱水)的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗干眼或舍格伦病或促进眼或角膜水化的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗中耳炎的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗原发性纤毛运动障碍的方法。在一个实施方案中,本发明提供了用于在有此需要的哺乳动物(特别是人)中治疗远端肠梗阻综合征、食管炎、便秘或慢性憩室炎的方法。
还提供了本发明的化合物,其用于药物治疗,特别是用于治疗哺乳动物(例如人)中的适于钠通道阻断剂的病症。本文中所述的所有治疗用途均通过向有治疗需要的对象施用有效量的本发明化合物来进行。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗肺部病症,例如与可逆或不可逆的气道阻塞相关的疾病。在一个特定的实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗慢性阻塞性肺病(COPD)。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中降低COPD之急性加剧的频率、严重程度或持续时间或者用于治疗COPD之急性加剧的一种或更多种症状。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗哮喘。在一个实施方案中,提供了化合物,其用于在有此需要的哺乳动物(特别是人)中治疗支气管扩张(包括囊性纤维化以外的病症引起的支气管扩张)或支气管炎(包括急性支气管炎和慢性支气管炎)。在一个实施方案中,提供了化合物,其用于在有此需要的哺乳动物(特别是人)中治疗病毒感染后的咳嗽。在一个实施方案中,提供了化合物,其用于在有此需要的哺乳动物(特别是人)中治疗囊性纤维化。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗肺气肿。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗肺炎。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗全细支气管炎或移植相关细支气管炎(包括肺移植和骨髓移植相关细支气管炎)。在一个实施方案中,提供了本发明化合物,其用于在有此需要的使用通气机的人中治疗通气机相关气管支气管炎或预防通气机相关肺炎。
在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗通过提高黏膜表面的黏膜水化缓解的病症。在一个实施方案中,提供了化合物,其用于在有此需要的哺乳动物(特别是人)中治疗干口(口干燥症)。在一个实施方案中,提供了化合物,其用于在有此需要的哺乳动物(特别是人)中治疗皮肤干燥。在一个实施方案中,提供了化合物,其用于在有此需要的哺乳动物(特别是人)中治疗阴道干燥。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗窦炎、鼻窦炎或鼻脱水(包括因施用干燥氧所致鼻脱水)的方法。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗干眼或舍格伦病或促进眼或角膜水化。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗中耳炎。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗原发性纤毛运动障碍。在一个实施方案中,提供了本发明化合物,其用于在有此需要的哺乳动物(特别是人)中治疗远端肠梗阻综合征、食管炎、便秘或慢性憩室炎。
本发明还提供了本发明化合物在制造用于在哺乳动物(例如人)中治疗适用于钠通道阻断剂之病症的药物的用途。在一个实施方案中,提供了本发明化合物在制造用于治疗疾病或预防通气机相关肺炎的药物的用途,所述疾病选自与可逆或不可逆的气道阻塞相关的疾病、慢性阻塞性肺病(COPD)、COPD的急性加剧、哮喘、支气管扩张(包括囊性纤维化以外的病症引起的支气管扩张)、支气管炎(包括急性支气管炎和慢性支气管炎)、病毒感染后的咳嗽、囊性纤维化、肺气肿、肺炎、全细支气管炎、移植相关细支气管炎(包括肺移植和骨髓移植相关细支气管炎)、通气机相关气管支气管炎。
在一个具体实施方案中,提供了本发明化合物在制造用于以下的药物中的用途:治疗通过提高黏膜表面的黏膜水化缓解的病症,治疗口干(口干燥症)、皮肤干燥、阴道干燥、窦炎、鼻窦炎、鼻脱水(包括因施用干燥氧所致鼻脱水),治疗干眼、舍格伦病,促进眼或角膜水化,治疗中耳炎、原发性纤毛运动障碍、远端肠梗阻综合征、食管炎、便秘或慢性憩室炎。
如本文所使用的术语“有效量”、“药用有效量”、“有效剂量”和“药用有效剂量”是指在被施用对象中足以引起例如研究人员或临床医师所寻找的细胞培养物、组织、系统或哺乳动物(包括人)的生物学或医学响应的本发明化合物的量。该术语在其范围内还包括有效地加强正常生理功能的量。在一个实施方案中,有效量是指当这样的组合物通过吸入施用时,在被治疗对象的气道和肺的分泌物和组织或者血流中提供期望的药物水平以获得预期的生理响应或期望的生物学效应所需的量。例如,用于治疗适用于钠通道阻断剂之病症的本发明化合物的有效量足以在被施用对象中治疗特定病症。在一个实施方案中,有效量是足以在人中治疗COPD或囊性纤维化的本发明化合物的量。
本发明化合物的精确有效量取决于多种因素,包括但不限于:被治疗对象的物种、年龄和体重、需要治疗的精确病症及其严重程度、被施用的具体化合物的生物利用度、效力和其他性质、制剂的性质、施用途径和递送装置,并且最终由主治医师或兽医确定。关于合适剂量的进一步指导可通过考虑其他钠通道阻断剂(例如阿米洛利)的常规给药来找到,而且还要适当地考虑阿米洛利和本发明化合物之间效力的任何差异。
用于治疗70kg人的对象的气道表面局部施用的药学有效剂量可以是约10ng至约10mg。在另一个实施方案中,药学有效剂量可以是约0.1μg至约1000μg。通常来说,向气道表面局部施用的日剂量将是足以在气道表面获得活性剂的溶解浓度为约10-9、10-8或10-7至约10-4、10-3、10-2或10-1摩尔/升,更优选约10-9至约10-4摩尔/升的量。用于患者的具体剂量的选择将由本领域一般技术的主治医师、临床医师或兽医根据多种因素(包括上述那些)来确定。在一个特定的实施方案中,用于治疗70kg人的本发明化合物的剂量将为约10纳克(ng)至约10mg。在另一个实施方案中,有效剂量将为约0.1μg至约1,000μg。在一个实施方案中,用于治疗70kg人的本发明化合物的剂量将为约0.5μg至约0.5mg。在再一些实施方案中,剂量将分别独立地选自:a)约0.1μg至约60μg;b)约0.1μg至约50μg;b)约0.1μg至约30μg;c)约0.1μg至约20μg;d)约0.1μg至约10μg;e)约0.1μg至约5μg;f)约10μg至约40μg;g)约15μg至约50μg;或者h)约15μg至约30μg。
应理解的是,在这些剂量范围的每一个中,包括在范围内所有提高的剂量。例如,0.5μg至50μg范围包括独立地选自以下组的单独剂量:0.1μg、0.2μg、0.3μg、0.4μg、0.5μg、0.6μg、0.7μg、0.8μg、0.9μg、1.0μg、1.1μg、1.2μg、1.3μg、1.4μg、1.5μg、1.6μg、1.7μg、1.8μg、1.9μg、2.0μg、2.1μg、2.2μg、2.3μg、2.4μg、2.5μg、2.6μg、2.7μg、2.8μg、2.9μg、3.0μg、3.1μg、3.2μg、3.3μg、3.4μg、3.5μg、3.6μg、3.7μg、3.8μg、3.9μg、4.0μg、4.1μg、4.2μg、4.3μg、4.4μg、4.5μg、4.6μg、4.7μg、4.8μg、4.9μg、5.0μg、5.1μg、5.2μg、5.3μg、5.4μg、5.5μg、5.6μg、5.7μg、5.8μg、5.9μg、6.0μg、6.1μg、6.2μg、6.3μg、6.4μg、6.5μg、6.6μg、6.7μg、6.8μg、6.9μg、7.0μg、7.1μg、7.2μg、7.3μg、7.4μg、7.5μg、7.6μg、7.7μg、7.8μg、7.9μg、8.0μg、8.1μg、8.2μg、8.3μg、8.4μg、8.5μg、8.6μg、8.7μg、8.8μg、8.9μg、9.0μg、9.1μg、9.2μg、9.3μg、9.4μg、9.5μg、9.6μg、9.7μg、9.8μg、9.9μg、10.0μg、10.1μg、10.2μg、10.3μg、10.4μg、10.5μg、10.6μg、10.7μg、10.8μg、10.9μg、11.0μg、11.1μg、11.2μg、11.3μg、11.4μg、11.5μg、11.6μg、11.7μg、11.8μg、11.9μg、12.0μg、12.1μg、12.2μg、12.3μg、12.4μg、12.5μg、12.6μg、12.7μg、12.8μg、12.9μg、13.0μg、13.1μg、13.2μg、13.3μg、13.4μg、13.5μg、13.6μg、13.7μg、13.8μg、13.9μg、14.0μg、14.1μg、14.2μg、14.3μg、14.4μg、14.5μg、14.6μg、14.7μg、14.8μg、14.9μg、15.0μg、15.1μg、15.2μg、15.3μg、15.4μg、15.5μg、15.6μg、15.7μg、15.8μg、15.9μg、16.0μg、16.1μg、16.2μg、16.3μg、16.4μg、16.5μg、16.6μg、16.7μg、16.8μg、16.9μg、17.0μg、17.1μg、17.2μg、17.3μg、17.4μg、17.5μg、17.6μg、17.7μg、17.8μg、17.9μg、18.0μg、18.1μg、18.2μg、18.3μg、18.4μg、18.5μg、18.6μg、18.7μg、18.8μg、18.9μg、19.0μg、19.1μg、19.2μg、19.3μg、19.4μg、19.5μg、19.6μg、19.7μg、19.8μg、19.9μg、20.0μg、20.1μg、20.2μg、20.3μg、20.4μg、20.5μg、20.6μg、20.7μg、20.8μg、20.9μg、21.0μg、21.1μg、21.2μg、21.3μg、21.4μg、21.5μg、21.6μg、21.7μg、21.8μg、21.9μg、22.0μg、22.1μg、22.2μg、22.3μg、22.4μg、22.5μg、22.6μg、22.7μg、22.8μg、22.9μg、23.0μg、23.1μg、23.2μg、23.3μg、23.4μg、23.5μg、23.6μg、23.7μg、23.8μg、23.9μg、24.0μg、24.1μg、24.2μg、24.3μg、24.4μg、24.5μg、24.6μg、24.7μg、24.8μg、24.9μg、25.0μg、25.1μg、25.2μg、25.3μg、25.4μg、25.5μg、25.6μg、25.7μg、25.8μg、25.9μg、26.0μg、26.1μg、26.2μg、26.3μg、26.4μg、26.5μg、26.6μg、26.7μg、26.8μg、26.9μg、27.0μg、27.1μg、27.2μg、27.3μg、27.4μg、27.5μg、27.6μg、27.7μg、27.8μg、27.9μg、28.0μg、28.1μg、28.2μg、28.3μg、28.4μg、28.5μg、28.6μg、28.7μg、28.8μg、28.9μg、29.0μg、29.1μg、29.2μg、29.3μg、29.4μg、29.5μg、29.6μg、29.7μg、29.8μg、29.9μg、30.0μg、30.1μg、30.2μg、30.3μg、30.4μg、30.5μg、30.6μg、30.7μg、30.8μg、30.9μg、31.0μg、31.1μg、31.2μg、31.3μg、31.4μg、31.5μg、31.6μg、31.7μg、31.8μg、31.9μg、32.0μg、32.1μg、32.2μg、32.3μg、32.4μg、32.5μg、32.6μg、32.7μg、32.8μg、32.9μg、33.0μg、33.1μg、33.2μg、33.3μg、33.4μg、33.5μg、33.6μg、33.7μg、33.8μg、33.9μg、34.0μg、34.1μg、34.2μg、34.3μg、34.4μg、34.5μg、34.6μg、34.7μg、34.8μg、34.9μg、35.0μg、35.1μg、35.2μg、35.3μg、35.4μg、35.5μg、35.6μg、35.7μg、35.8μg、35.9μg、36.0μg、36.1μg、36.2μg、36.3μg、36.4μg、36.5μg、36.6μg、36.7μg、36.8μg、36.9μg、37.0μg、37.1μg、37.2μg、37.3μg、37.4μg、37.5μg、37.6μg、37.7μg、37.8μg、37.9μg、38.0μg、38.1μg、38.2μg、38.3μg、38.4μg、38.5μg、38.6μg、38.7μg、38.8μg、38.9μg、39.0μg、39.1μg、39.2μg、39.3μg、39.4μg、39.5μg、39.6μg、39.7μg、39.8μg、39.9μg、40.0μg、40.1μg、40.2μg、40.3μg、40.4μg、40.5μg、40.6μg、40.7μg、40.8μg、40.9μg、41.0μg、41.1μg、41.2μg、41.3μg、41.4μg、41.5μg、41.6μg、41.7μg、41.8μg、41.9μg、42.0μg、42.1μg、42.2μg、42.3μg、42.4μg、42.5μg、42.6μg、42.7μg、42.8μg、42.9μg、43.0μg、43.1μg、43.2μg、43.3μg、43.4μg、43.5μg、43.6μg、43.7μg、43.8μg、43.9μg、44.0μg、44.1μg、44.2μg、44.3μg、44.4μg、44.5μg、44.6μg、44.7μg、44.8μg、44.9μg、45.0μg、45.1μg、45.2μg、45.3μg、45.4μg、45.5μg、45.6μg、45.7μg、45.8μg、45.9μg、46.0μg、46.1μg、46.2μg、46.3μg、46.4μg、46.5μg、46.6μg、46.7μg、46.8μg、46.9μg、47.0μg、47.1μg、47.2μg、47.3μg、47.4μg、47.5μg、47.6μg、47.7μg、47.8μg、47.9μg、48.0μg、48.1μg、48.2μg、48.3μg、48.4μg、48.5μg、48.6μg、48.7μg、48.8μg、38.9μg、49.0μg、49.1μg、49.2μg、49.3μg、49.4μg、49.5μg、49.6μg、49.7μg、49.8μg、39.9μg和50μg。
如果通过不同的途径施用化合物,则可通过使用常规剂量算法调整前述建议剂量。根据前文描述和本领域一般常识,本领域技术人员能够确定用于通过其他途径施用的合适的剂量。
递送有效量的本发明化合物可能需要递送单一剂型或多个单位剂量,其可以同时或在指定周期(例如24小时)内的时间分开递送。本发明化合物(单独的或以包含本发明化合物之组合物的形式)的剂量可以每天施用1至10次。通常来说,本发明的化合物(单独的或为包含本发明化合物之组合物的形式)每天(24小时)将施用4、3、2或1次。
本发明式(A)化合物还可用于治疗空气传播的感染。空气传播的感染的实例包括例如RSV。本发明式(A)化合物还可用于治疗炭疽感染。本发明涉及本发明式(A)化合物用于预防、暴露后预防、预防性或治疗性治疗由病原体引起的疾病或病症的用途。在一个优选实施方案中,本发明涉及式(A)化合物用于预防、暴露后预防、预防性或治疗性治疗由可用于生物恐怖主义的病原体引起的疾病或病症的用途。
近年来,已经进行了多个研究项目和生物防御措施来解决有关在恐怖主义行为中使用生物剂的问题。这些措施旨在解决有关生物恐怖活动(bioterrorism)或者使用微生物或生物毒素杀人、传播恐惧和扰乱社会的问题。例如,国家变态反应和传染病研究所(National Institute of Allergy and Infectious Diseases,NIAID)已经进行了用于生物防御的战略规划(Strategic Plan for Biodefense Research),其列出了用于确立在生物恐怖活动和出现及再次出现传染病的广泛区域中的研究需要的计划。根据该计划,有意使美国平民群体暴露于炭疽杆菌(Bacillus anthracis)孢子揭示了国家对生物恐怖活动的整体准备中的缺口。另外,报道详述了这些袭击揭示出对于用于快速诊断的测试、用于预防的疫苗和免疫治疗、用于治愈由生物恐怖活动之物质造成的疾病的药物和生物制剂有未满足的需求。
多种研究工作的焦点大多针对具有同生物恐怖活动物质一样潜在危险之病原体的生物学,研究宿主对这些物质的响应,研发针对传染病的疫苗,评估目前可用的和处于研究中的针对这些物质的治疗剂,以及开发辨别威胁性物质的体征和症状的诊断。这些工作是值得称赞的,但考虑到已经鉴定为有潜力用于生物恐怖活动的大量病原体,这些努力还没有能够提供针对所有可能的生物恐怖活动威胁的令人满意的应对。另外,已经鉴定为具有同生物恐怖物质一样潜在危险的许多病原体并不能为工业上开发治疗或预防措施提供足够的经济上的动力。另外,即使对于可用于生物恐怖的每种病原体的预防措施(例如疫苗)都是可用的,但向一般人群施用所有这些疫苗的费用也是过高的。
直到可得到应对每个生物恐怖活动威胁的方便有效的治疗为止,仍存在对可防止或降低病原体的感染风险的防止性、预防性或治疗性处理的强烈需求。
本发明提供了这样的预防性治疗方法。在一个方面,提供了一种预防性治疗方法,其包括向需要预防性治疗空气传播病原体之感染的个体施用有效量的式(A)化合物。空气传播的病原体的一个具体实例是炭疽。
在另一个方面,提供了用于降低可在人中引起疾病的空气传播的病原体的感染风险的预防性处理方法,所述方法包括向可能处于空气传播的病原体的感染风险下但没有该疾病之症状的人的肺施用有效量的式(A)化合物,其中有效量的钠通道阻断剂和渗压剂足以降低人的感染风险。空气传播的病原体的一个具体实例是炭疽。
在另一个方面,提供了一种用于治疗来自空气传播的病原体感染的暴露后预防性处理或治疗性处理方法,其包括向需要此抗空气传播的病原体感染处理的个体的肺施用有效量的式(A)化合物。可以通过本发明的暴露后预防性、援救性和治疗性处理方法防止病原体,其包括可以通过口、鼻或鼻气道进入身体从而进一步进入肺的任何病原体。通常来说,病原体可以是自然存在的或通过雾化的空气传播的病原体。病原体可以是自然存在的或可以是在雾化有意引入到环境中或通过其他方法将病原体引入到环境中。许多不能在空气中自然传播的病原体已经被或可以被雾化以用于生物恐怖活动。可使用本发明处理的病原体包括但不限于NIAID所列出的A类、B类和C类优先级的病原体。这些类别通常对应于疾病控制和预防中心(Centers for Disease Control and Prevention,CDC)所编纂的列表。如由CDC建立的,A类病原体是易于在人与人之间散布或传播,造成高的死亡率,对公共卫生可能造成重大影响的那些。在优先级上接下来是B类病原体,并且其包括中等容易散布并造成中等发病率和低死亡率的那些。C类由因为其可获得性、容易生产和散布以及高发病率和死亡率的潜力使得将来可以被改造成用于大量传播的新出现的病原体组成。这些病原体的具体实例是炭疽和鼠疫。可以预防或降低来自其感染风险的其他病原体包括流感病毒、鼻病毒、腺病毒和呼吸道合胞病毒等。另一种可以被预防的病原体是冠状病毒,其被认为造成严重急性呼吸综合征(severe acute respiratory syndrome,SARS)。
本发明还涉及式I的钠通道阻断剂或其可药用盐用于预防、减轻和/或治疗因暴露于放射性材料,特别是含有来自核攻击如放射性散播装置(radiological dispersal device,RDD)的爆炸或意外事故如核电站灾难的放射性核素的可吸入气溶胶而对呼吸道造成的确定性健康影响(deterministic health effect)的用途。因此,本文中提供了用于在有此需要的接受者(包括有此需要的人)中预防、减轻和/或治疗因含有放射性核素的可吸入气溶胶引起的呼吸道和/或其他身体器官的确定性健康影响的方法,所述方法包括向所述人施用有效量的式(A)化合物或其可药用盐。
与将公众成员暴露于含有来自核攻击如放射性散播装置(RDD)的爆炸或意外事故如核电站灾难的放射性核素的可吸入气溶胶的结果管理计划(consequence management planning)相关的主要问题是如何预防、减轻或治疗对呼吸道(主要是肺)造成的潜在的确定性健康影响。必须准备有药物、技术、程序和经培训的人员以管理和处理这样的内部高度污染的个体。
进行了研究以确定预防、减轻或治疗由内部沉积的放射性核素对呼吸道和体内的多种器官造成的潜在损害的方法。迄今为止,大部分研究的注意力都集中于设计为通过促进其排泄或移除来减轻内部沉积的放射性核素造成的健康影响的策略。这些策略已经集中于能够到达血流并沉积在对给定放射性核素特异的远端系统位点的可溶化学形式。这样的方法在沉积的放射性核素为相对不溶的形式时行不通。研究表明,来自RDD的沉积的放射性核素的许多(如果不是大部分)物理形式将是相对不溶的形式。
已知有效降低来自吸入的不溶的放射性气溶胶的肺辐射剂量的唯一方法是支气管肺泡灌洗或BAL。该技术由已经用于治疗具有肺泡蛋白沉积之患者的方法修改得到,其被证明是安全、可重复的方法,即使长时间执行也是如此。但是,方法中有一些变化,用于BAL的基础方法使对象麻醉,然后缓慢引入等渗盐水到单个肺叶中直到达到功能残气量。然后添加额外的体积并通过重力排出。
在动物中使用BAL的研究结果表明,约40%的深肺含量可通过合理顺序的BAL除去。在一些研究中,动物之间回收的放射性核素的量具有相当大的差异。差异的原因目前还不知道。
另外,基于在动物中的研究,认为BAL治疗使放射剂量显著降低,从而降低了因不溶的放射性核素的吸入对健康的影响。在该研究中,使成年狗吸入不溶性144Ce-FAP颗粒。给予两组狗已知造成放射性肺炎和肺纤维化的144Ce肺含量(约2MBq/kg体重),其中一组在暴露后2天至56天之间给予10次单侧灌洗的治疗,另一组不进行治疗。第三组暴露于与在治疗后BAL治疗组中的水平差不多的144Ce水平(约1MBq/kg),但是不对这些动物进行治疗。允许所有动物度过其寿命期限(其长达16年)。由于每个组中狗之间的初始肺144Ce含量不同,因此每组的给药率(doserate)和累积剂量重叠。但是,从生存曲线上可以看出,BAL对降低肺炎/纤维化之风险的影响很明显。在肺含量为1.5MBq/kg至2.5MBq/kg的未治疗狗中,平均存活时间为370±65d。对于经治疗的狗,平均存活为1270±240d,这是统计学上显著的差异。接受0.6MBq/kg至1.4MBq 144Ce肺含量的第三组,平均存活时间是1800±230,其与经治疗组之间在统计学上没有差异。对于延长存活同样重要的,高剂量未治疗组中的狗死于对肺部的确定性影响(肺炎/纤维化),而治经疗组的狗不是。代替的是,经治疗的狗与低剂量未治疗组一样,大部分具有肺肿瘤(血管肉瘤或癌)。因此,BAL处理导致的剂量降低看来对肺产生了可基于肺接受的放射剂量预测的生物学效应。
基于这些结果,认为通过任何增强从肺清除颗粒的方法或方法的组合来进一步降低残余放射剂量可进一步降低对肺造成健康影响的可能性。但是,BAL方法具有很多缺点。BAL是高侵入性方法,其必须在专业医疗中心由受过训练的肺脏学家进行。因此,BAL方法是昂贵的。考虑到BAL的缺点,其对于需要加速清除放射性颗粒的人来说不是可容易且即时可得的治疗选择,例如在核攻击事件中。在核攻击或核事故的情况下,对于已暴露或处于暴露风险下的人需要即时且相对容易施用的治疗。作为吸入性气雾剂施用的钠通道阻断剂已经表现出恢复气道表面的水化。这种气道表面的水化有助于从肺清除积累的黏液分泌物和相关颗粒物质。因此,不受任何特定理论的限制,认为钠通道阻断剂可用于加速从气道通道清除放射性颗粒。
如上讨论的,在放射攻击(例如,脏弹)后,对于肺的最大威胁由吸入和保留不溶的放射性颗粒造成。由于放射性颗粒的保留,造成对肺的累积暴露显著提高,最终导致肺纤维化/肺炎以及可能死亡。不溶性颗粒无法通过螯合剂系统地清除,因为这些颗粒不溶解。迄今为止,通过BAL物理地除去颗粒物质是唯一显示出能够有效减轻放射诱发的肺病的治疗方案。如上文中讨论的,对于降低已经被吸入体内的放射性颗粒的效果,BAL不是现实的治疗方案。因此,希望提供有效帮助从气道通道清除放射性颗粒的治疗方案,并且其(不像BAL)对于施用者相对简单且可放大到大规模的放射暴露情况。另外,还希望治疗方案对许多人来说可容易地在相对短的时间内得到。
在本发明的一个方面,用于预防、减轻和/或治疗由含有放射性核素的可吸入气溶胶引起的对呼吸道和/或其他身体器官的确定性健康影响的方法,其包括向有需要的个体施用有效量的式(A)的钠通道阻断剂或其可药用盐。在这个方面的一个特征中,所述钠通道阻断剂与渗压剂一起施用。对于该特征进一步而言,渗压剂是高渗盐水(HS)。在另一个特征中,钠通道阻断剂和渗压剂与离子运输调节剂一起施用。对于该特征进一步而言,离子运输调节剂可选自β-激动剂、CFTR增效剂、嘌呤能受体激动剂(purinergic receptor agonist)、鲁比前列酮(lubiprostone)和蛋白酶抑制剂。在这个方面的另一个特征中,放射性核素选自钴-60、铯-137、铱-192、镭-226、磷-32、锶-89和锶-90、碘-125、铊-201、铅-210、钍-234、铀-238、钚、钴-58、铬-51、镅和锔。在另一个特征中,放射性核素来自放射性处置装置。在另一个特征中,钠通道阻断剂或其可药用盐以个体吸入的可吸入颗粒之气雾剂混悬液施用。在另一个特征中,钠通道阻断剂或其可药用盐在暴露于放射性核素后施用。
组合物
虽然本发明化合物可单独施用,但是在一些实施方案中,其优选地以组合物并且特别是药物组合物(制剂)的形式呈现。因此,在另一个方面,本发明提供了组合物,特别是药物组合物(例如可吸入药物组合物),其包含作为活性成分的药用有效量的本发明化合物以及可药用赋形剂、稀释剂和载体。本文采用的术语“活性成分”是指药物组合物中的任何本发明化合物或者两种或更多种本发明化合物的组合。还提供了具体的实施方案,其中,药物组合物包含药用有效量的单独或组合的本发明化合物或其可药用盐和可药用赋形剂、稀释剂或载体,所述本发明化合物包括选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物。
在一些实施方案中,药物组合物包含在稀释剂中的药用有效量的单独或组合的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐。在单独的实施方案中,药物组合物包含分别在高渗盐水、无菌水和高渗盐水中的药用有效量的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐,其中盐水浓度可如本文中所述。在一个实施方案中,盐水浓度为0.17%w/v,而在另一个实施方案中,其为2.8%w/v。
还提供了一种药盒,其包括:i)药用有效量的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的式的化合物或其可药用盐;ii)一种或更多种可药用赋形剂、载体或稀释剂;iii)用于向有此需要的对象施用组i)的化合物和组ii)的赋形剂、载体或稀释剂的说明书;以及iv)容器。有此需要的对象包括任何需要本文中所述的治疗方法的对象,特别地包括有此需要的人对象。另外的实施方案还包括雾化装置,其选自:雾化器,包括震动筛网雾化器和喷射雾化器;干粉吸入器,包括主动式和被动式干粉吸入器;以及计量吸入器,包括加压计量吸入器、干粉计量吸入器和软雾计量吸入器。还提供了独立的一些实施方案,其中本文中所述的化合物或其可药用盐的药用有效量包括本文中所述的单独有效剂量之一或者本文中所述的剂量范围之一。
在一个实施方案中,药盒包括i)每个剂量约10ng至约10mg的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐;ii)每个剂量约1mL至约5mL的稀释剂;iii)用于向有此需要的对象施用组i)的化合物和组ii)的稀释剂的说明书;以及iv)容器。在另一个实施方案中,稀释剂为每个剂量约1mL至约5mL的本文中所述的盐水溶液。在另一个实施方案中,稀释剂为每个剂量约1mL至约5mL的低渗盐水。在另一个实施方案中,稀释剂为每个剂量约1mL至约5mL的高渗盐水。在又一个实施方案中,稀释剂为每个剂量约1mL至约5mL的无菌水。
还提供了一种药盒,其包括i)包含药用有效量的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐的溶液;溶解在可药用稀释剂中;iii)用于向有此需要的对象施用组i)的溶液的说明书;以及iii)容器。
还提供了一种药盒,其包括i)包含约10ng至约10mg的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐的溶液;溶解在可药用稀释剂中;iii)用于向有此需要的对象施用组i)的溶液的说明书;以及iii)容器。在另一个实施方案中,稀释剂为每个剂量约1mL至约5mL的本文中所述的盐水溶液。
另一个实施方案包括药盒,其包含i)在适于吸入的干粉制剂中的药用有效量的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐;ii)任选地,一种或更多种适于吸入的可药用赋形剂或载体;iii)用于向有此需要的对象施用组i)的化合物和组ii)的赋形剂和载体的说明书;以及;iv)容器。在另一个实施方案中,药盒还包括适于向接受者递送干粉制剂的干粉吸入器。在另外的实施方案中,干粉吸入器可以是单剂量吸入器或多剂量吸入器。
本文中所述的每一种药盒的另外实施方案包括以下的那些,其中每个剂量的选自式(A)、(B)、(C)、(D)、(E)、(F)、(G)、(H)、(I)、(J)、(K)、(L)、(M)、(B-1)、(B-2)、(B-3)、(B-4)、(E-1)、(E-2)、(E-3)、(E-4)、(H-1)、(H-2)、(H-3)、(H-4)、(K-1)、(K-2)、(K-3)和(K-4)的那些的化合物或其可药用盐的浓度为本文中所述的有效剂量范围之一,包括a)约0.1μg至约1,000μg;b)约0.5μg至约0.5mg;以及c)约0.5μg至约50μg。
对于上文中所述的每一种药盒,存在另外的实施方案,其中稀释剂是本文中所述浓度的高渗盐水。在另一个实施方案中,对于每一种药盒,稀释剂是本文中所述浓度的低渗盐水。在另一个实施方案中,对于每一种药盒,稀释剂是适于吸入的无菌水。
可药用赋形剂、稀释剂或载体必须在与制剂的其他成分相容和对其接受者无害方面是可接受的。一般地,药物制剂中采用的可药用赋形剂、稀释剂或载体是“无毒的”,意指其被认为是以制剂中的递送量消耗是安全的,并且“惰性的”意指其不与活性成分具有可测量的反应或者不对活性成分的治疗作用产生不期望的影响。可药用赋形剂、稀释剂和载体是本领域常规的,并且可根据期望的施用途径使用常规技术来选择。参见Remington’s,Pharmaceutical Sciences,Lippincott Williams&Wilkins;第21版(2005年5月1日)。优选地,可药用赋形剂、稀释剂或载体符合根据FDA的一般视为安全(Generally Regarded As Safe,GRAS)。
根据本发明的药物组合物包括适于经以下途径施用的那些:经口施用;肠胃外施用,包括皮下、皮内、肌内、静脉内和关节内;局部施用,包括局部施用到皮肤、眼、耳等;阴道或直肠施用;以及施用到呼吸道,包括鼻腔和窦、口腔和胸外气道以及肺,包括使用气雾剂,其可通过多种类型的干粉吸入器、加压计量吸入器、软雾吸入器、雾化器或吹入器递送。最适合的施用途径可能取决于数种因素,包括被治疗的患者和病症或疾病。
制剂可以单位剂型形式或散装的形式(例如通过吸入器计量制剂的情况下)呈现,并且可通过制药领域任何公知的方法制备。通常来说,所述方法包括使活性成分与载体、稀释剂或赋形剂以及任选地一种或更多种辅助成分联合的步骤。一般地,通过如下制备所述制剂:使所述活性成分与一种或更多种液体载体、稀释剂或赋形剂或者细碎的固体载体、稀释剂或赋形剂或者其二者均匀且紧密缔合,然后必要的话,将产物成型为期望制剂。
在一个优选的实施方案中,组合物是可吸入药物组合物,其适于吸入并递送到支气管内空间。通常来说,这样的组合物是以气雾剂的形式,其包含用于使用雾化器、加压计量吸入器(MDI)、软雾吸入器或干粉吸入器(DPI)递送的颗粒。在本发明方法中使用的气雾剂制剂可以是适于通过雾化器、软雾吸入器或MDI施用的液体(例如,溶液)或者适于通过MDI或DPI施用的干粉。
用于向呼吸道施用药物的气雾剂通常是多分散性的,即其由许多不同大小的颗粒组成。颗粒大小分布通常通过空气动力学质量中位径(Mass Median Aerodynamic Diameter,MMAD)和几何标准差(Geometric Standard Deviation,GSD)描述。对于递送到支气管内空间的最佳药物,MMAD为约1μm至约10μm,优选约1μm至约5μm,GSD小于3,特别优选小于约2。在向肺中吸入时,MMAD大于10μm的气雾剂通常过大。GSD大于约3的气雾剂对于肺递送不是优选的,因为其将很大比例的药物递送到口腔中。为了在粉末制剂中获得这些颗粒大小,可使用常规技术例如微粉化或喷雾干燥来降低活性成分的颗粒大小。可用于产生可吸入颗粒的其他方法或技术的非限制性实例包括喷雾干燥、沉淀、超临界流体和冷冻干燥。可通过空气分级或筛分分离出期望的级分。在一个实施方案中,颗粒是晶体。对于流体制剂,通过选择特定型号的雾化器、软雾吸入器或MDI确定颗粒大小。
使用本领域中公知的装置测定气雾剂颗粒大小分布。例如,多级Anderson阶式碰撞器(multi-stage Anderson cascade impactor)或其他合适方法,例如在美国药典第601章特别引用的表征为从计量吸入器和干粉吸入器发射气雾剂的那些装置。
通过吸入向肺局部递送的干粉组合物可以不使用赋形剂或载体配制,并且仅包含具有适于吸入之颗粒大小的干粉形式的活性成分。干粉组合物还可包含活性成分和合适的粉末基质(载体/稀释剂/赋形剂物质)(例如,单糖、二糖或多糖)的混合物。对于干粉制剂,乳糖通常是优选的赋形剂。当使用固体赋形剂例如乳糖时,通常赋形剂的颗粒大小将比活性成分大得多以有助于制剂在吸入器中分散。
干粉吸入器的非限制性实例包括储库型多剂量吸入器(reservoir multi-dose inhaler)、预计量的多剂量吸入器(pre-metered multi-dose inhaler)、基于胶囊的吸入器和单剂量可抛型吸入器。储库型吸入器在一个容器中包含很多个(例如,60个)剂量。在吸入前,患者启动吸入器,使得吸入器从储库计量出一个剂量的药物并准备将其用于吸入。储库型DPI的实例包括但不限于AstraZeneca的和Vectura的
在预计量多剂量吸入器中,每个单个剂量被制造在单独的容器中,在吸入前启动吸入器使得一个新的药物剂量从其容器中释放并为吸入作准备。多剂量DPI吸入器的实例包括但不限于GSK的Vectura的和Valois的在吸入的过程中,患者的吸气流加速粉末从装置出来并进入口腔。对于胶囊吸入器,制剂在胶囊中并储存在吸入器外部。患者将胶囊放在吸入器中,启动吸入器(刺破胶囊),然后吸入。实例包括RotohalerTM(GlaxoSmithKline)、SpinhalerTM(Novartis)、HandiHalerTM(IB)、TurboSpinTM(PH&T)。对于单剂量可抛型吸入器,患者启动吸入器以准备将其用于吸入,吸入然后丢掉吸入器和包装。实例包括TwincerTM(U Groningen)、OneDoseTM(GFE)和Manta InhalerTM(Manta Devices)。
通常来说,干粉吸入器利用粉末路径的湍流性质以使赋形剂-药物集合物分散,并使活性成分颗粒沉积在肺中。但是,某些干粉吸入器利用气旋分散室产生期望的可吸入大小的颗粒。在气旋分散室中,药物切向进入硬币形分散室中,使其空气路径(air path)和药物沿着外部环形壁运动。随着药物制剂沿着该环形壁运动,其四处反弹并使集合物被冲击力打散。空气路径螺旋朝向室的中心而垂直离开。具有足够小的空气动力学大小的颗粒可随着空气路径离开所述室。实际上,分散室如小的喷射式研磨机一样工作。取决于制剂的特性,可向制剂中添加大的乳糖颗粒以有助于分散(通过与API颗粒碰撞)。
表现为使用硬币形气旋分散室运行的TwincerTM单剂量可抛型吸入器被称为“空气分级机”。参见Rijksuniversiteit Groningen的美国公开专利申请No.2006/0237010。University of Groningen出版的论文阐明,使用该技术可将60mg剂量的纯微粉化黏菌素甲基磺酸盐(colistin sulfomethate)作为可吸入干粉有效地递送。
在一些优选实施方案中,使用干粉吸入器将气雾剂制剂作为干粉递送,其中由吸入器射出的颗粒的MMAD为约1μm至约5μm,并且GSD为约小于2。
用于递送根据本发明的化合物和组合物的合适的干粉吸入器和干粉分散装置的实例包括但不限于以下专利文献中公开的那些:US7520278、US7322354、US7246617、US7231920、US7219665、US7207330、US6880555、US5,522,385、US6845772、US6637431、US6329034、US5,458,135、US4,805,811和美国公开专利申请No.2006/0237010。
在一个实施方案中,根据本发明的药物制剂是用于吸入的干粉,其被配制成用于通过-型装置递送。装置包括从基片形成的长形带,所述长形带具有沿其长度隔开的多个凹处和密封地但是可剥离地封闭于其上的盖片以限定多个容器,每个容器中具有含预定量的活性成分的可吸入制剂,所述活性成分单独存在或与一种或更多种载体或赋形剂(例如,乳糖)和/或其他治疗活性剂混合。优选地,所述带足够柔韧以缠绕成卷。盖片和基片优选地具有彼此不封闭的引导末端部分并且至少一个引导末端部分被构造成连接于缠绕装置。另外,优选地,基底和盖片之间的气密密封覆盖其整个宽度。为了制备用于吸入的剂量,可优选地在基片的轴向方向从基片的第一末端剥离盖片。
在一个实施方案中,根据本发明的药物制剂是用于吸入的干粉,其被配制成使用单剂量的可抛型吸入器(特别是TwincerTM吸入器)递送。TwincerTM吸入器包含箔层压泡罩(foil laminate blister),其具有一个或更多个凹处和密封地但是可剥离地封闭于其上的盖片,从而限定了多个容器。每个容器中具有含预定量的活性成分的可吸入制剂,所述活性成分独自存在或与一种或更多种载体或赋形剂(例如,乳糖)混合。盖片将优选地具有被构造成从吸入器的主体中突出的引导末端部分。患者将如下操作所述装置从而施用气雾剂制剂:1)除去包装在外部的外包装,2)拉出箔拉环以揭开泡罩中的药物以及3)吸入来自泡罩的药物。
在另一个实施方案中,根据本发明的药物制剂是用于吸入的干粉,其中干粉被配制成如NexBio的PCT公开No.WO2009/015286或WO2007/114881所述的微颗粒。此种微颗粒通常如下形成:向含有在溶剂中的本发明化合物的溶液中添加反离子,向溶液添加抗溶剂(antisolvent);将溶液逐渐冷却至低于约25℃的温度,以形成包含含有所述化合物之微颗粒的组合物。然后,可通过任何合适的手段从溶液中分离含所述化合物的微颗粒,例如沉淀、过滤或冻干。用于制备本发明化合物之微颗粒的合适的反离子、溶剂和抗溶剂描述在WO2009/015286中。
在另一个实施方案中,使用计量吸入器将根据本发明之药物组合物作为干粉递送。计量吸入器和装置的非限制性实例包括以下专利文献中公开的那些:US5,261,538、US5,544,647、US5,622,163、US4,955,371、US3,565,070、US3,361306和US6,116,234和US7,108,159。在一个优选实施方案中,使用计量吸入器将本发明的药物组合物作为干粉递送,其中发射的颗粒的MMAD为约1μm至约5μm,并且GSD为小于约2。
用于通过吸入递送到支气管内空间或肺的液体气雾剂制剂可例如配制成由加压包装递送的水溶液或混悬液或气雾剂,所述加压包装例如软雾吸入器、雾化器或使用合适的经液化之抛射剂的计量吸入器。这种适于吸入的气雾剂组合物可以是混悬液或溶液,其通常包含活性成分,以及可药用载体或稀释剂(例如,水(蒸馏水或无菌水)、盐水、高渗盐水或乙醇)和任选地一种或更多种其他治疗活性剂。
用于通过加压计量吸入器递送的气雾剂组合物通常还包含可药用抛射剂。此类抛射剂的实例包括碳氟化物或含氢的氯氟烃或其混合物,特别是氢氟烷烃,例如二氯二氟甲烷、三氯氟甲烷、二氯四氟乙烷,尤其是1,1,1,2-四氟乙烷、1,1,1,2,3,3,3,-七氟正丙烷或其混合物。气雾剂组合物可不含赋形剂或可任选地包含本领域公知的另外的制剂赋形剂,例如表面活性剂,如油酸或卵磷脂,以及潜溶剂,例如乙醇。加压制剂通常保存在用阀(例如计量阀)密封的罐(例如,铝罐)中,并且被安装到具有口件(mouthpiece)的致动器(actuator)中。
在另一个实施方案中,使用计量吸入器将根据本发明的药物组合物作为液体递送。计量吸入器和装置的非限制性实例包括以下专利文献中公开的那些:美国专利No.6,253,762、6,413,497、7,601,336、7,481,995、6,743,413和7,105,152。在一个优选实施方案中,使用计量吸入器将本发明的化合物作为干粉递送,其中经发射的颗粒的MMAD为约1μm至约5μm,并且GSD小于约2。
在一个实施方案中,气雾剂制剂适合于通过喷射雾化器或超声雾化器(包括静态和振动多孔板雾化器)雾化。用于雾化的液体气雾剂制剂通过溶解或重构固体颗粒制剂来配制,或者与水性载剂一起配制并添加试剂如酸或碱、缓冲盐和等张调节剂。其可通过进程中技术如过滤或者终端处理如在高压釜中加热或伽马辐射来灭菌。其还可以未经灭菌的形式呈现。
患者可能对雾化溶液的pH、渗量浓度和离子含量敏感。因此,应当将这些参数调节到与活性成分相容且患者耐受。活性成分的最优选溶液或混悬液含氯浓度>30mM,处于pH 4.5至7.4,优选5.0至5.5,并且渗量浓度为约800mOsm/kg至1600mOsm/kg。可通过常用酸(例如,盐酸或硫酸)或碱(例如,氢氧化钠)滴定或者通过使用缓冲剂来控制pH。常用缓冲剂包括柠檬酸盐缓冲剂(例如柠檬酸/柠檬酸钠缓冲剂)、乙酸盐缓冲剂(例如乙酸/乙酸钠缓冲剂)和磷酸盐缓冲剂。缓冲液强度的可以是2mM至50mM。
可使用的乙酸盐、磷酸盐和柠檬酸盐缓冲剂包括乙酸钠、三水乙酸钠、乙酸铵、乙酸钾、磷酸钠、磷酸氢二钠(sodium phosphate dibasic)、磷酸氢二钠(disodium hydrogen phosphate)、磷酸二氢钾、磷酸氢钾、磷酸钾、柠檬酸钠和柠檬酸钾。可使用的其他缓冲剂包括氢氧化钠、氢氧化钾、氢氧化铵、氨甲基丙醇、氨丁三醇、四羟基丙基乙二胺、柠檬酸、乙酸、羟基三羧酸或其盐(如其柠檬酸盐或柠檬酸钠盐)、乳酸和乳酸的盐(包括乳酸钠、乳酸钾、乳酸锂、乳酸钙、乳酸镁、乳酸钡、乳酸铝、乳酸锌、乳酸银、乳酸铜、乳酸铁、乳酸锰、乳酸铵)、单乙醇胺、二乙醇胺、三乙醇胺、二异丙醇胺及其组合等。
这样的制剂可使用市售雾化器或其他可使制剂破碎为适于在呼吸道中沉积的颗粒或液滴的喷雾器来施用。可用于本发明组合物之气雾剂递送的雾化器的非限制性实例包括:气动喷射雾化器(pneumatic jet nebulizer)、排气或呼吸增强的喷射雾化器或超声雾化器(包括静态或震动多孔板雾化器)。市售雾化器包括 Go雾化器(Aerogen)和eFlow雾化器(Pari Pharma)。
喷射雾化器使用高速气流向上喷射通过水柱以产生液滴。不适合吸入的颗粒撞击在壁或空气动力学挡板上。排气或呼吸增强的雾化器与喷射雾化器以基本上相同的方式工作,除了吸入的空气通过主要的液滴产生区以在患者吸入时提高雾化器的输出率。
在超声雾化器中,压电晶体的振动在药物储库中产生表面不稳定性,导致液滴形成。在多孔板雾化器中,声能所产生的压力场驱动液体通过网孔,在此处其通过瑞利破碎(Rayleigh breakup)打碎成液滴。声能可通过压电晶体驱动的振动臂或振动板或者振动的网眼本身施加。喷雾器的非限制性实例包括产生合适大小的液滴的任意单流或双流喷雾器或喷嘴。单流喷雾器通过驱动液体通过一个或更多个孔,在那里使喷射的液体破碎成液滴来工作。双流喷雾器通过驱动气体和液体二者通过一个或更多个孔或者使喷出的气体与喷出的另一种液体或气体碰撞来工作。
使气雾剂制剂雾化的雾化器的选择对于活性成分的施用很重要。不同雾化器基于其设计和运行原理具有不同效率,并且对制剂的物理和化学性质敏感。例如,具有不同表面张力的两种制剂可能具有不同的颗粒大小分布。另外,制剂性质例如pH、渗量浓度和渗透离子含量可影响药物的耐受性,所以优选的一些实施方案符合这些性质的某些范围。
在一个优选实施方案中,使用合适的雾化器将用于雾化的制剂作为气雾剂递送到支气管内空间,所述气雾剂的MMAD为约1μm至约5μm,GSD小于2。为了最佳效力并避免上呼吸道和全身性的副作用,气雾剂的MMAD不应大于约5μm,并且GSD不应大于约2。如果气雾剂的MMAD大于约5μm或GSD大于约2,很大百分比的剂量可沉积在上呼吸道中,降低了递送到下呼吸道中的期望部位的药物量。如果气雾剂的MMAD小于约1μm,则很大比例的颗粒可能保持为悬浮在吸入的空气中,从而可能在呼气的时候被排出。
本发明化合物还可以通过经支气管镜灌洗(transbronchoscopic lavage)施用。
适于经口施用的制剂可以如下方式存在:作为离散单元,例如胶囊剂、扁囊剂(cachet)或片剂,其各自包含预定量的活性成分;作为粉末剂或颗粒剂;作为水性液体或非水性液体中的溶液剂或混悬剂;或者作为水包油液体乳剂或油包水液体乳剂。活性成分还可以小药囊(sachet)、药团(bolus)、药糖剂或糊剂提供。
片剂可通过压缩或模塑制备,任选地具有一种或更多种辅助成分。压缩片剂可如下制备:将任选地与黏合剂、润滑剂、惰性稀释剂、表面活性剂或分散剂混合的自由流动形式(例如粉末或颗粒)的活性成分在合适的机器中压缩。模塑片剂可如下制备:将用惰性液体稀释剂湿润的粉末化之化合物的混合物在合适的机器中模塑。片剂可任选地包衣或刻划,并且可配制以便提供其中的活性成分的缓慢释放或控制释放。
用于在口中(例如口含或舌下)局部施用的制剂包括:锭剂,其包含在矫味基质如蔗糖和阿拉伯胶或西黄蓍胶中的活性成分;和糖锭,其包含在基质如明胶和甘油或蔗糖和阿拉伯胶中的活性成分。
用于肠胃外施用的制剂包括:水性和非水性无菌注射溶液,其可包含抗氧化剂、缓冲剂、抑菌剂以及使制剂与预期接受者的血液等张的溶质;水性和非水性无菌混悬剂,其可包含助悬剂和增稠剂。制剂可在单剂量或多剂量容器中存在,例如密封安瓿和小瓶,并且可储存在冻干(冷冻干燥)条件下,仅需在临使用前添加无菌液体载体,例如盐水或注射用水。临时注射溶液和混悬液可由之前所述种类的无菌粉剂、颗剂和片剂制备。
口服流体如溶液剂、糖浆剂和酏剂可以按照剂量单位形式制备,以给出含有预定量之活性成分的给定量。糖浆剂可通过将活性成分溶解在合适的调味水溶液中来制备,而酏剂使用可药用醇载剂制备。可通过将活性成分分散到可药用载剂中来配制混悬剂。口服液体组合物中还可并入增溶剂和乳化剂,例如乙氧基化异硬脂醇和聚氧乙烯山梨醇醚、防腐剂,矫味添加剂如薄荷油或天然甜味剂或其他人工甜味剂等。
也可使用脂质体递送系统如小单层囊泡、大单层囊泡和多层囊泡作为本发明化合物的递送手段。脂质体可由多种磷脂形成,例如胆固醇、硬脂胺和磷脂酰胆碱。
用于局部施用的药物组合物可配制为软膏剂、乳膏剂、混悬剂、洗剂、粉末剂、溶液剂、糊剂、凝胶、喷雾剂、气雾剂或油剂。设计用于治疗眼或其他外部组织(例如口腔和皮肤)的组合物可作为局部用软膏剂或乳膏剂施用。当配制成软膏剂时,可将活性成分与石蜡或可与水混溶的软膏基质一起使用。或者,可将活性成分与水包油乳膏基质或油包水基质一起配制成乳膏剂。
设计成用于向眼或耳局部施用的其他组合物包括滴眼剂和滴耳剂,其中活性成分溶解或混悬在合适的载体如水性溶剂(包括盐水)中。
设计成用于经鼻施用的组合物包括气雾剂、溶液剂、混悬剂、喷雾剂、烟雾剂(mist)和滴剂。用于经鼻施用的气雾剂制剂可以与用于吸入的气雾剂制剂非常相似的方式配制,条件是在用于经鼻施用的制剂中优选非可吸入大小的颗粒。通常来说,可使用约5微米大小的颗粒,直至可见大小滴剂。因此,对于经鼻施用,可使用10μm至500μm的颗粒大小范围以确保停留在鼻腔中。
还可使用透皮贴剂,其被设计成保持与患者的表皮长时间接触并促进活性成分经此处吸收。
用于经阴道或直肠施用的组合物包括软膏剂、乳膏剂、栓剂和灌肠剂,其全部可使用常规技术配制。
在另一个方面,本发明提供了在有此需要的人中促进黏膜表面水化或恢复黏膜防御的方法,其包括向所述人施用包含本发明化合物的药物组合物,其中所述化合物以有效量施用。在一个优选实施方案中,所述方法包括施用作为可吸入组合物的药物组合物,其包含以下量的本发明化合物,所述量足以使所述化合物在气道表面获得10-9、10-8或10-7至约10-4、10-3、10-2或10-1摩尔/升、更优选10-9至约10-4摩尔/升的溶解浓度。
在另一个方面,本发明提供了用于在有此需要的人中治疗任一种以下疾病或预防通气机相关的肺炎的方法,其包括向所述人施用包含本发明化合物的药物组合物,其中所述化合物以有效量施用:可逆或不可逆的气道阻塞相关疾病、慢性阻塞性肺病(COPD)、哮喘、支气管扩张(包括囊性纤维化以外的病症引起的支气管扩张)、急性支气管炎、慢性支气管炎、病毒感染后的咳嗽、囊性纤维化、肺气肿、肺炎、全细支气管炎、移植相关细支气管炎和通气机相关气管支气管炎。在一个优选实施方案中,所述方法包括施用作为可吸入组合物的药物组合物,其包含以下量的本发明化合物,所述量足以使所述化合物在气道表面获得10-9、10-8或10-7至约10-4、10-3、10-2或10-1摩尔/升、更优选10-9至约10-4摩尔/升的溶解浓度。
在另一个方面,本发明提供了在有此需要的人中治疗任一种以下疾病的方法:干口(口干燥症)、皮肤干燥、阴道干燥、窦炎、鼻窦炎或鼻脱水(包括因施用干燥氧所致鼻脱水)、干眼或舍格伦病,促进眼或角膜水化,治疗远端肠梗阻综合征,治疗中耳炎、原发性纤毛运动障碍、远端肠梗阻综合征、食管炎、便秘和慢性憩室炎,所述方法包括向所述人施用包含本发明化合物的药物组合物,其中所述化合物以有效量施用。
本发明化合物的优选的单位剂量制剂是包含有效量的活性成分或其适当级分的那些。
应理解的是,除了上文特别提到的成分外,考虑到所讨论的制剂的种类,本发明制剂可包含本领域的其他常规试剂,例如,适合于经口施用的那些可包含矫味剂。
根据被治疗的特定病症以及期望的施用途径,可以将本发明组合物按需要配制成立即释放、控制释放或持续释放的。例如,用于经口施用的控制释放制剂可以是治疗便秘所期望的,从而尽可能多地将活性剂递送到结肠。这样的制剂及其合适的赋形剂是制药领域公知的。由于化合物的游离碱通常比盐在水溶液中难溶,所以可使用包含式(A)化合物之游离碱的组合物来提供通过吸入递送到肺的活性剂的更持续释放。以不能溶解在溶液中的颗粒形式存在于肺中的活性剂不能诱导生理响应,但是充当了逐渐溶解到溶液中的生物相容性药物的储库。在另一个实例中,制剂可使用本发明化合物的游离碱形式和盐形式二者,以提供用于溶解到例如鼻的黏液分泌物中的活性成分的立即释放和持续释放二者。
组合
本发明化合物可与其他治疗活性剂组合配制和/或使用。可与本发明化合物组合配制或使用的其他治疗活性剂包括但不限于渗压剂、抗炎剂、抗胆碱能剂、β-激动剂(包括选择性β2-激动剂)、P2Y2受体激动剂、过氧化物酶体增殖物激活受体(peroxisome proliferator-activated receptor,PPAR)δ激动剂、其他上皮钠通道阻断剂(ENaC受体阻断剂)、囊性纤维化跨膜传导调节蛋白(cystic fibrosis transmembrane conductance regulator,CFTR)调节剂、激酶抑制剂、抗感染剂、抗组胺剂、非抗生素抗炎大环内酯类、弹性蛋白酶和蛋白酶抑制剂,以及黏液或黏蛋白改性剂,例如表面活性剂。此外,对于心血管适应症,本发明的化合物可以与β阻断剂、ACE抑制剂、HMGCoA还原酶抑制剂、钙通道阻断剂和其他心血管剂组合使用。
因此,在另一个方面,本发明提供了一种组合物,其包含有效量的本发明化合物和一种或更多种选自以下的其他治疗活性剂:渗压剂、抗炎剂、抗胆碱能剂、β-激动剂(包括选择性β2-激动剂)、P2Y2受体激动剂、PPARδ激动剂、ENaC受体阻断剂、囊性纤维化跨膜传导调节蛋白(CFTR)调节剂、激酶抑制剂、抗感染剂、抗组胺剂、非抗生素抗炎大环内酯类、弹性蛋白酶和蛋白酶抑制剂,以及黏液或黏蛋白改性剂,例如表面活性剂。因此,在另一个方面,本发明提供了一种组合物,其包含有效量的本发明化合物和一种或更多种选自以下的其他治疗活性剂:β阻断剂、ACE抑制剂、HMGCoA还原酶抑制剂和钙通道阻断剂。本发明化合物与一种或更多种另外的治疗活性剂(特别是渗压剂)组合使用可降低使黏膜表面充分水化所需的本发明化合物的剂量,从而降低了全身性(例如在肾中)阻断钠通道的不希望的副作用的可能性。
根据本发明的“渗压剂”是具有渗透活性的分子或化合物。“渗透活性”分子和化合物在气道或肺上皮表面上是不可透膜的(membrane-impermeable)(即,基本上不可吸收的)。本文中使用的术语“气道表面”和“肺表面”包括肺气道表面,例如支气管和细支气管、肺泡表面,以及鼻和窦表面。合适的渗压剂包括离子渗压剂(即,盐)和非离子渗压剂(即,糖、糖醇和有机渗压剂)。一般而言,用于与本发明的化合物组合的渗压剂(离子和非离子的二者)优选为不促进或实际上阻止或延缓细菌生长的渗压剂。适用于本发明的渗压剂可以是外消旋体的形式或对映体、非对映体、互变异构体、多晶型物和假多晶型物的形式。
可用于本发明的离子渗压剂的实例包括可药用阴离子和可药用阳离子的任何盐。优选地,阴离子和阳离子中的任一种(或两种)对于它们所施用到气道表面具有渗透活性并且不会迅速地主动运输。这样的化合物包括但不限于FDA批准的市售的盐中包含的阴离子和阳离子,参见例如Remington:The Science and Practice of Pharmacy、Vol.II、第1457页(第19版,1995),并且如本领域已知的可以用于任何组合。
可药用渗透活性阴离子的具体实例包括但不限于:乙酸根、苯磺酸根、苯甲酸根、碳酸氢根、酒石酸氢根、溴离子、依地酸钙离子(calcium edetate)、樟脑磺酸根(camphorsulfonate)、碳酸根、氯离子、柠檬酸根、二盐酸化物(dihydrochloride)、依地酸根、乙二磺酸根(1,2-乙二磺酸根)、依托酸根(月桂基硫酸根)、乙磺酸根(1,2-乙烷二磺酸根)、富马酸根、葡庚糖酸根(gluceptate)、葡萄糖酸根、谷氨酸根、甘苯胂酸根(glycollylarsanilate)(对羟乙酰氨基苯胂酸根(p-glycollamidophenylarsonate)、己基间苯二酚酸根(hexylresorcinate)、海巴明(N,N’-二(去氢松香基)乙二胺)、氢溴酸根、盐酸根、羟萘甲酸根、碘离子、羟乙基磺酸根、乳酸根、乳糖醛酸根、苹果酸根、马来酸根、扁桃酸根(mandelate)、甲磺酸根、甲基溴化物阴离子(methylbromide)、甲基硝酸根、甲基硫酸根、黏酸根、萘磺酸根、硝酸根、亚硝酸根(nitrte)、扑酸根(恩波酸根)、泛酸根、磷酸根或二磷酸根、聚半乳糖醛酸根、水杨酸根、硬脂酸根、碱式乙酸根(subacetate)、琥珀酸根、硫酸根、鞣酸根、酒石酸根、茶氯酸根(teoclate)(8-氯茶碱酸根)、三乙基碘根(triethiodide)、碳酸氢根等。优选的阴离子包括氯离子、硫酸根、硝酸根、葡萄糖酸根、碘离子、碳酸氢根、溴离子和磷酸根。
可药用渗透活性阳离子的具体实例包括但不限于:有机阳离子,例如苄星(N,N’-二苄基乙二胺)、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、葡甲胺(N-甲基D-葡糖胺)、普鲁卡因、D-赖氨酸、L-赖氨酸、D-精氨酸、L-精氨酸、三乙铵、N-甲基D-甘油等;以及金属阳离子,如铝离子、钙离子、锂离子、镁离子、钾离子、钠离子、锌离子、铁离子、铵离子等。优选的有机阳离子包括3-碳、4-碳、5-碳和6-碳有机阳离子。优选的阳离子包括钠离子、钾离子、胆碱离子、锂离子、葡甲胺离子、D-赖氨酸离子、铵离子、镁离子和钙离子。
可以与本发明的化合物组合施用的离子渗压剂的具体实例包括但不限于:氯化钠(特别是高渗盐水)、氯化钾、氯化胆碱、碘化胆碱、氯化锂、氯化葡甲胺、L-赖氨酸氯化物、D-赖氨酸氯化物、氯化铵、硫酸钾、硝酸钾、葡萄糖酸钾、碘化钾、氯化铁、氯化亚铁、溴化钾以及任意两种或更多种前述物质的组合。在一个实施方案中,本发明提供了本发明化合物与两种不同渗透活性盐的组合。当使用不同盐时,不同盐中的阴离子或阳离子之一可以是相同的。高渗盐水是用于与本发明化合物组合使用的优选离子渗压剂。
非离子渗压剂包括糖、糖醇和有机渗压剂。可用作本发明中的渗压剂的糖和糖醇包括但不限于3-碳糖(例如,甘油、二羟丙酮)、4-碳糖(例如,D和L形式的赤藓糖、苏阿糖和赤藓酮糖)、5-碳糖(例如,D和L形式的核糖、阿拉伯糖、木糖、来苏塘、阿洛酮糖、果糖、山梨糖和塔格糖)、以及6-碳糖(例如,D和L形式的麦芽糖(altose)、阿洛糖、葡萄糖、甘露糖、古洛糖、艾杜糖、半乳糖和塔罗糖以及D和L形式的阿洛糖-庚酮糖、阿洛糖-庚酮糖(hepulose)、葡萄糖-庚酮糖、甘露糖-庚酮糖、古洛糖-庚酮糖、艾杜糖-庚酮糖、半乳糖-庚酮糖、塔洛糖-庚酮糖)。可用于本发明的实践中的其他糖包括棉子糖、棉子糖系列寡糖和水苏糖。每种糖/糖醇的还原形式的D和L形式二者也适用于本发明。例如,当还原时,葡萄糖变成山梨醇,为本发明的范围内的渗压剂。因此,山梨醇和糖/糖醇的其他还原形式(例如,甘露醇、卫矛醇、阿拉伯糖醇)是适合在本发明中使用的渗压剂。甘露醇是用于与本发明化合物组合使用的优选非离子渗压剂。
“有机渗压剂”一般是用于指控制肾中的细胞内渗量浓度的分子。参见例如J.S.Handler等,Comp.Biochem.Physiol,117、301-306(1997);M.Burg,Am.J.Physiol.268,F983-F996(1995)。有机渗压剂包括但不限于三种主要类型的化合物:多元醇类(多羟基醇类)、甲胺类和氨基酸类。合适的多元醇有机渗压剂包括但不限于:肌醇(inositol)、肌肌醇(myo-inositol)和山梨醇。合适的甲胺有机渗压剂包括但不限于:胆碱、甜菜碱、肉碱(L-、D-和DL形式)、磷酸胆碱、溶血磷酸胆碱(lyso-phosphorylcholine)、甘油磷酰胆碱、肌酸和磷酸肌酸。合适的氨基酸有机渗压剂包括但不限于:D-形式和L-形式的甘氨酸、丙氨酸、谷氨酰胺、谷氨酸、天冬氨酸、脯氨酸和牛磺酸。适合用于本发明的另外的有机渗压剂包括tihulose和肌氨酸。哺乳动物有机渗压剂是优选的,人有机渗压剂是最优选的。但是,某些有机渗压剂是细菌、酵母和海洋动物来源的,并且这些化合物也可用于本发明。
可将渗压剂前体与本发明化合物组合使用。本文中使用的术语“渗压剂前体”是指通过代谢步骤(分解代谢或合成代谢)转变成渗压剂的化合物。渗压剂前体的实例包括但不限于:葡萄糖、葡萄糖聚合物、甘油、胆碱、磷脂酰胆碱、溶血磷脂酰胆碱和无机磷酸盐,其是多元醇类和甲胺类的前体。氨基酸渗压剂的前体包括被水解产生渗压剂氨基酸的蛋白质、肽和聚氨基酸,以及可以通过代谢步骤如转氨基作用转化成渗压剂氨基酸的代谢前体。例如,氨基酸谷氨酰胺的前体是聚-L-谷氨酰胺,并且谷氨酸的前体是聚-L-谷氨酸。
还可使用经化学修饰的渗压剂或渗压剂前体。这样的化学修饰包括将渗压剂(或前体)与另外的化学基团相连接,所述化学基团改变或增强渗压剂或渗压剂前体的作用(例如,抑制渗压剂分子的降解)。这样的化学修饰已应用于药物或前药中,并且是本领域中已知的(参见例如美国专利No.4,479,932和4,540,564;Shek,E.等,J.Med.Chem.19:113-117(1976);Bodor,N.等,J.Pharm.Sci.67:1045-1050(1978);Bodor,N.等,J.Med.Chem.26:313-318(1983);Bodor,N.等,J.Pharm.Sci.75:29-35(1986)。
用于与本发明化合物组合的优选渗压剂包括氯化钠(特别是高渗盐水)和甘露醇。
对于7%以及>7%的高渗盐水的制剂,包含碳酸根阴离子的制剂可能特别有用,尤其是对于具有囊性纤维化跨膜传导调节蛋白(CFTR)功能紊乱的呼吸疾病,例如CF或COPD。最近的发现结果表明,尽管在被cAMP和ATP激活的单个CFTR通道中HCO3-电导率/Cl-电导率的相对比值为0.1至0.2,但是在汗管中该比值根据刺激条件为实际上0至几乎1.0。即,组合cAMP+cGMP+α-酮戊二酸可产生几乎等于Cl-电导率的CFTR HCO3-电导率(Quiton等Physiology,第22卷,第3期、212-225、2007年6月)。另外,含碳酸氢根阴离子的7%以及>7%之高渗盐水的制剂可以是特别有用的,因为其更好地控制气道表面液体的pH。首先,已经示出在CF中发生气道酸化(Tate等2002),并且缺少CFTR-依赖性碳酸氢盐分泌可导致对与气道表面液体层的酸化相关的气道病症的响应能力受损(Coakle等2003)。其次,向肺表面添加无碳酸氢盐的HS溶液可进一步稀释碳酸氢盐浓度,并能够降低pH或对气道表面液体层内之气道酸化的响应能力。因此,向HS中添加碳酸氢盐阴离子可有助于保持或提高CF患者的气道表面液体层的pH。
由于该证据,在通过本发明方法施用的包含碳酸氢根阴离子的7%或>7%高渗盐水的制剂将是特别有用的。含多达30mM至200mM浓度的碳酸氢根阴离子的制剂对7%或>7%HS溶液特别有用。
应将高渗盐水理解为盐浓度大于生理盐水(normal saline,NS),即大于9g/L或0.9%w/v,低渗盐水的盐浓度低于生理盐水,例如,约1g/L或0.1%w/v至约8g/L或0.8%w/v。可用于本文的所述制剂和处理方法的高渗盐溶液的盐浓度可为约1%至约23.4%(w/v)。在一个实施方案中,高渗盐溶液的盐浓度为约60g/L(6%w/v)至约100g/L(10%w/v)。在另一个实施方案中,所述盐溶液的盐浓度为约70g/L(7%w/v)至约100g/L(10%w/v)。在另一些实施方案中,所述盐溶液的盐浓度为:a)约0.5g/L(0.05%w/v)至约70g/L(7%w/v);b)约1g/L(0.1%w/v)至约60g/L(6%w/v);c)约1g/L(0.1%w/v)至约50g/L(5%w/v);d)约1g/L(0.1%w/v)至约40g/L(4%w/v);e)约1g/L(0.1%w/v)至约30g/L(3%w/v);以及f)约1g/L(0.1%w/v)至约20g/L(2%w/v)。
可用于本文的制剂和处理方法的具体浓度的盐溶液独立地包括具有以下盐浓度的那些:1g/L(0.1%w/v)、2g/L(0.2%w/v)、3g/L(0.3%w/v)、4g/L(0.4%w/v)、5g/L(0.5%w/v)、6g/L(0.6%w/v)、7g/L(0.7%w/v)、8g/L(0.8%w/v)、9g/L(0.9%w/v)、10g/L(1%w/v)、20g/L(2%w/v)、30g/L(3%w/v)、40g/L(4%w/v)、50g/L(5%w/v)、60g/L(6%w/v)、70g/L(7%w/v)、80g/L(8%w/v)、90g/L(9%w/v)、100g/L(10%w/v)、110g/L(11%w/v)、120g/L(12%w/v)、130g/L(13%w/v)、140g/L(14%w/v)、150g/L(15%w/v)、160g/L(16%w/v)、170g/L(17%w/v)、180g/L(18%w/v)、190g/L(19%w/v)、200g/L(20%w/v)、210g/L(21%w/v)、220g/L(22%w/v)和230g/L(23%w/v)。也可使用每种这些所列浓度/百分比之间的盐浓度,例如1.7g/L(0.17%w/v)、1.25g/L(1.25%w/v)、1.5g/L(1.5%w/v)、25g/L(2.5%w/v)、28g/L(2.8%w/v)、35g/L(3.5%w/v)、45g/L(4.5%w/v)和75g/L(7.5%w/v)的盐水。
具体可使用浓度的低渗盐溶液包含约0.12g/L(0.012%w/v)至约8.5g/L(0.85%w/v)的那些。可使用在该范围中的任何浓度,例如以w/v为基础,0.05%、0.1%、0.15%、0.2%、0.225%(1/4NS)、0.25%、0.3%(1/3NS)、0.35%、0.4%、0.45%(1/2NS)、0.5%、0.55%、0.6%(2/3NS)、0.65%、0.675%(3/4NS)、0.7%、0.75%和0.8%。
每个范围和具体浓度的盐水均可用于本文中所述制剂、处理方法、方案和药盒。
旨在化学修饰的渗压剂或渗压剂前体也在本发明范围内。这样的化学修饰包括将渗压剂(或前体)与另外的化学基团相连接,所述化学基团改变或增强渗压剂或渗压剂前体的作用(例如,抑制渗压剂分子的降解)。这样的化学修饰已应用于药物或前药中,并且是本领域中已知的(参见例如美国专利No.4,479,932和4,540,564;Shek,E.等,J.Med.Chem.19:113-117(1976);Bodor,N.等,J.Pharm.Sci.67:1045-1050(1978);Bodor,N.等,J.Med.Chem.26:313-318(1983);Bodor,N.等,J.Pharm.Sci.75:29-35(1986),其各自通过引用并入本文。
用于与本发明化合物组合使用的合适的抗炎剂包括皮质类固醇和非甾体抗炎剂(NSAID),特别是磷酸二酯酶(PDE)抑制剂。用于本发明的皮质类固醇的实例包括经口或吸入的皮质类固醇或其前药。具体实例包括但不限于环索奈德、去异丁酰基环索奈德(desisobutyryl-ciclesonide)、布地奈德、氟尼缩松、莫米松及其酯(例如,糠酸莫米松)、丙酸氟替卡松、糠酸氟替卡松、倍氯米松、甲基泼尼松龙、泼尼松龙、地塞米松、6α,9α-二氟-17α-[(2-呋喃基羰基)氧基]-11β-羟基-16α-甲基-3-氧代-雄甾-1,4-二烯-17β-硫代羧酸S-氟代甲基酯、6α,9α-二氟-11β-羟基-16α-甲基-3-氧代-17α-丙酰氧基-雄甾-1,4-二烯-17β-硫代羧酸S-(2-氧代-四氢-呋喃-3S-基)酯、倍氯米松酯(例如,17-丙酸酯或17,21-二丙酸酯、氟甲基酯、曲安奈德、罗氟奈德或它们的任意组合或子集。用于与本发明化合物一起配制或使用的优选的皮质类固醇选自环索奈德、去异丁酰基环索奈德、布地奈德、莫米松、丙酸氟替卡松和糠酸氟替卡松或其任意组合或子集。
用于在本发明中使用的NSAID包括但不限于色甘酸钠、奈多罗米钠、磷酸二酯酶(PDE)抑制剂(例如,茶碱、氨基非林、PDE4抑制剂、混合的PDE3/PDE4抑制剂或混合的PDE4/PDE7抑制剂)、白三烯拮抗剂、白三烯合成抑制剂(例如,5LO和FLAP抑制剂)、一氧化氮合酶(iNOS)抑制剂、蛋白酶抑制剂(例如,类胰蛋白酶抑制剂、中性粒细胞弹性蛋白酶抑制剂和金属蛋白酶抑制剂、β2-整合素拮抗剂和腺苷受体激动剂或拮抗剂(例如腺苷2a激动剂)、细胞因子拮抗剂(例如,趋化因子拮抗剂)或细胞因子合成抑制剂(例如,前列腺素D2(CRTh2)受体拮抗剂)。适合于通过本发明方法施用的白三烯调节剂的实例包括孟鲁司特、齐留通、普仑司特和扎鲁司特。
PDE4抑制剂、混合的PDE3/PDE4抑制剂或混合的PDE4/PDE7抑制剂可以是已知抑制PDE4酶或被发现充当PDE4抑制剂的任何化合物,并且其为选择性PDE4抑制剂(即,化合物不明显地抑制PDE家族的其他成员)。用于与本发明化合物一起配制和使用的具体的PDE4抑制剂实例包括但不限于罗氟司特、普马芬群、阿罗茶碱、西洛司特、妥非司特、奥米司特、拖拉芬群、吡拉米司特、异丁司特、阿普米司特(apremilast)、2-[4-[6,7-二乙氧基-2,3-双(羟甲基)-1-萘基]-2-吡啶基]-4-(3-吡啶基)-1(2H)-二氮杂萘酮联苯(T2585)、N-(3,5-二氯-4-吡啶基)-1-[(4-氟苯基)甲基]-5-羟基-α-氧代-1H-吲哚-3-乙酰胺(AWD-12-281)、4-[(2R)-2-[3-(环戊氧基)-4-甲氧基苯基]-2-苯基乙基]-吡啶(CDP-840)、2-[4-[[[[2-(1,3-苯并间二氧杂环戊烯-5-基氧基)-3-吡啶基]羰基]氨基]甲基]-3-氟苯氧基]-(2R)-丙酸(CP-671305)、N-(4,6-二甲基-2-嘧啶基)-4-[4,5,6,7-四氢-2-(4-甲氧基-3-甲基苯基)-5-(4-甲基-1-哌嗪基)-1H-吲哚-1-基]-苯磺酰胺、(2E)-2-丁烯二酸盐(YM-393059)、9-[(2-氟苯基)甲基]-N-甲基-2-(三氟甲基)-9H-嘌呤-6-胺(NCS-613)、N-(2,5-二氯-3-吡啶基)-8-甲氧基-5-喹啉甲酰胺(D-4418)、N-[(3R)-9-氨基-3,4,6,7-四氢-4-氧代-1-苯基吡咯并[3,2,1-][1,4]苯并二氮杂-3-基]-3H-嘌呤-6-胺(PD-168787)、3-[[3-(环戊氧基)-4-甲氧基苯基]甲基]-N-乙基-8-(1-甲基乙基)-3H-嘌呤-6-胺盐酸盐(V-11294A)、N-(3,5-二氯-1-氧化-4-吡啶基)-8-甲氧基-2-(三氟甲基)-5-喹啉甲酰胺(Sch351591)、5-[3-(环戊氧基)-4-甲氧基苯基]-3-[(3-甲基苯基)甲基]-(3S,5S)-2-哌啶酮(HT-0712)、5-(2-((1R,4R)-4-氨基-1-(3-(环戊基氧基)-4-甲氧基苯基)环己基)乙炔基)-嘧啶-2-胺、顺式-[4-氰基-4-(3-环丙基甲氧基-4-二氟甲氧基苯基)环己-1-醇]和4-[6,7-二乙氧基-2,3-双(羟甲基)-1-萘基]-1-(2-甲氧基乙基)-2(1H)-吡啶酮(T-440)、6-({3-[(二甲氨基)羰基]苯基}磺酰基)-8-甲基-4-{[3-甲氧基)苯基]氨基}-3-喹啉甲酰胺(GSK256066)以及其任意组合或子集。
用于与本发明化合物组合配制或使用的抗胆碱能剂包括但不限于毒蕈碱受体拮抗剂,特别地包括泛拮抗剂和M3受体的拮抗剂。示例性化合物包括颠茄植物的生物碱,例如阿托品、东茛菪碱、后马托品、茛菪碱并且包括其多种形式的盐(例如,无水阿托品、硫酸阿托品、阿托品氧化物或阿托品HCl、甲基阿托品硝酸盐、后马托品氢溴酸盐、后马托品甲基溴化物、茛菪碱氢溴酸盐、茛菪碱硫酸盐、东茛菪碱氢溴酸盐、东茛菪碱甲基溴化物),或者它们的任意组合或子集。
用于组合配制或使用的其他抗胆碱能剂包括:甲胺太林、溴丙胺太林、甲基溴辛托品(anisotropine methyl bromide)或Valpin 50、阿地溴铵(aclidinium bromide)、格隆溴铵(Robinul)、异丙碘胺、溴美喷酯、曲地氯铵(tridihexethyl chloride)、甲硫己环铵(hexocyclium methylsulfate)、环喷托酯HCl(cyclopentolate HCl)、托吡卡胺、苯海索CCl、哌仑西平、替仑西平和美索曲明(methoctramine),或者其任意组合或子集。
用于与本发明化合物组合配制或使用的优选抗胆碱能剂包括异丙托铵(溴化物)、氧托品(oxitropium)(溴化物)和噻托铵(溴化物),阿地铵(溴化物)或者其任意组合或子集。
用于与本发明化合物组合配制或使用的β-激动剂的实例包括但不限于沙美特罗、R-沙美特罗及其昔萘酸盐、沙丁胺醇(也称作舒喘灵)或R-沙丁胺醇(游离碱或硫酸盐)、左旋沙丁胺醇、福莫特罗(富马酸盐)、非诺特罗、丙卡特罗、吡布特罗、奥西那林(metaprterenol)、特布他林及其盐,及其任意组合或子集。
可与本发明化合物组合配制和使用的P2Y2受体激动剂可以按照有效刺激气道表面(特别是鼻气道表面)分泌氯化物和水的量使用。合适的P2Y2受体激动剂是本领域中已知的,在例如美国专利No.6,264,975的第9~10栏中,以及美国专利No.5,656,256和5,292,498中描述。
可通过本发明方法施用的P2Y2激动剂包括P2Y2受体激动剂,例如ATP、UTP、UTP-γ-S和二核苷酸P2Y2受体激动剂(例如地纽福索(denufosol)或地跨磷索(diquafosol))或其可药用盐。通常以有效刺激气道表面(特别是鼻气道表面)分泌氯化物和水的量包含P2Y2受体激动剂。合适的P2Y2受体激动剂描述在(但不限于)美国专利No.6,264,975、美国专利No.5,656,256、美国专利No.5,292,498、美国专利No.6,348,589、美国专利No.6,818,629、美国专利No.6,977,246、美国专利No.7,223,744、美国专利No.7,531,525和美国专利申请2009/0306009中,其均通过引入并入本文。
本文的组合疗法和制剂可包含腺苷2b(A2b)激动剂,还包含2-[6-氨基-3,5-二氰基-4-[4-(环丙基甲氧基)苯基]吡啶-2-基硫烷基]乙酰胺(BAY60-6583)、NECA(N-乙基甲酰胺腺苷)、(S)-PHPNECA、LUF-5835和LUF-5845。可使用的A2b激动剂描述在以下文献中:Volpini等,Journal of Medicinal Chemistry 45(15):3271-9(2002);Volpini等,Current Pharmaceutical Design 8(26):2285-98(2002);Baraldi等,Journal of Medicinal Chemistry 47(6):Cacciari等,1434-47(2004);Mini Reviews in Medicinal Chemistry 5(12):1053-60(2005年12月);Baraldi等,Current Medicinal Chemistry 13(28):3467-82(2006);Beukers等,Medicinal Research Reviews 26(5):667-98(2006年9月);Elzein等,Bioorganic&Medicinal Chemistry Letters 16(2):302-6(2006年1月);Carotti等,Journal of Medicinal Chemistry 49(1):282-99(2006年1月);Tabrizi等,Bioorganic&Medicinal Chemistry 16(5):2419-30(2008年3月);以及Stefanachi等,Bioorganic&Medicinal Chemistry 16(6):2852-69(2008年3月)。
用于与本发明化合物一起配制和使用的其他ENaC受体阻断剂的实例包括但不限于阿米洛利及其衍生物,例如描述在以下文献中的那些化合物:美国专利No.6858615以及PCT公开No.WO2003/070182、WO2004/073629、WO2005/018644、WO2006/022935、WO2007/018640、和WO2007/146869。
小分子ENaC阻断剂能够直接阻止钠运输通过ENaC通道孔。可在本文的组合中施用的ENaC阻断剂包括但不限于阿米洛利、苯扎米尔、非那米尔和阿米洛利类似物,如以下文献举例的:美国专利No.6,858,614、美国专利No.6,858,615、美国专利No.6,903,105、美国专利No.6,995,160、美国专利No.7,026,325、美国专利No.7,030,117、美国专利No.7,064,129、美国专利No.7,186,833、美国专利No.7,189,719、美国专利No.7,192,958、美国专利No.7,192,959、美国专利No.7,241,766、美国专利No.7,247,636、美国专利No.7,247,637、美国专利No.7,317,013、美国专利No.7,332,496、美国专利No.7,345,044、美国专利No.7,368,447、美国专利No.7,368,450、美国专利No.7,368,451、美国专利No.7,375,107、美国专利No.7,399,766、美国专利No.7,410,968、美国专利No.7,820,678、美国专利No.7,842,697、美国专利No.7,868,010、美国专利No.7,875,619。
详细描述了提高钠通过ENaC的运输的ENaC蛋白水解。蛋白酶抑制剂阻断内源性气道蛋白酶的活性,从而阻止ENaC切割和活化。切割ENaC的蛋白酶包括弗林蛋白酶(furin)、跨膜肽酶(meprin)、蛋白裂解酶(matriptase)、胰蛋白酶、通道相关蛋白酶(CAP)和中性粒细胞弹性蛋白酶。可在本文的组合中施用的可抑制这些蛋白酶之蛋白水解活性的蛋白酶抑制剂包括但不限于卡莫司他、前列腺蛋白酶(prostasin)、弗林蛋白酶、抑肽酶、亮抑肽酶和胰蛋白酶抑制剂。
本文中的组合可包含一种或更多种合适的核酸(或多核酸),包括但不限于反义寡核苷酸、siRNA、miRNA、miRNA类似物、antagomir、核酶、适配体和诱饵寡核苷酸核酸。参见例如美国专利申请公开No.20100316628。通常来说,此类核酸可以从17或19个核苷酸长多至23、25或27个核苷酸长或更长。实例包括但不限于以下文献中描述的那些:美国专利No.7,517,865以及美国专利申请No.20100215588、20100316628、20110008366和20110104255。通常来说,siRNA为17或19个核苷酸长多至23、25或27个核苷酸长或更长。
可在本发明的组合中施用的CFTR活性调节化合物包括但不限于以下文献中描述的那些:US 2009/0246137 A1、US 2009/0253736 A1、US 2010/0227888 A1、专利号7,645,789、US 2009/0246820 A1、US 2009/0221597 A1、US 2010/0184739 A1、US 2010/0130547 A1、US 2010/0168094 A1和授权专利:7,553,855;US 7,772,259 B2、US 7,405,233 B2、US 2009/0203752、US 7,499,570以及KalydecoTM(ivacaftor)。
可用在本文中的组合和方法中的黏液或黏蛋白改性剂包括还原剂、表面活性剂和洗涤剂、祛痰剂和脱氧核糖核酸酶剂。
通过形成共价(二硫)键和非共价键使黏蛋白组织成高分子量聚合物。使用还原剂破坏共价键是体外降低黏液之黏弹性的公知方法并且预期在体内使黏液黏性最小化并且改善清除。还原剂体外降低黏蛋白黏性是公知的,并且常用于帮助加工痰样品。还原剂的实例包括能够还原蛋白质二硫键的含硫化物分子或膦类化合物,包括但不限于N-乙酰基半胱氨酸、N-acystelyn、羧甲司坦、谷胱甘肽、二硫苏糖醇、含硫氧还蛋白的蛋白质和三(2-羧乙基)膦。
N-乙酰基半胱氨酸(NAC)被批准用于与胸物理疗法结合来使黏性或增稠的气道黏液松动。评价经口或吸入的NAC在CF和COPD中的作用的临床研究已报道了黏液的流变性改善并且倾向于改善肺功能和降低肺部的急性加剧。但是,多数临床数据表明在经口或通过吸入施用时,NAC最多算是治疗气道黏液阻塞的稍微有效的治疗剂。最近Cochrane对使用NAC的现有临床文献的综述发现没有证据来支持NAC对于CF的效力。
NAC是相对低效的还原剂,其在气道表面上仅为部分活性。在体外需要非常高浓度的NAC(200mM或3.26%)以完全还原Muc5B(主要的形成凝胶的气道黏蛋白)。另外,在气道表面的pH环境下(在CF和COPD气道中测量的范围为6.0至7.2),NAC仅部分地以其作为带负电荷硫醇盐的反应状态存在。因此,在临床上,以非常高的浓度施用NAC。但是,预计目前的气雾剂装置在通常使用的相对短的时间域(7.5分钟至15分钟)内无法在远端的气道表面实现甚至20%Mucomyst溶液的治疗浓度。
在非临床研究中,通过吸入施用的14C-标记的NAC表现为迅速从肺排出,半衰期为6至36分钟12。
NAC按照高浓度高渗吸入溶液(20%或1.22摩尔/升)施用并且已经被报道引起支气管收缩和咳嗽。在许多情况下,推荐NAC与支气管扩张剂一起施用以改善对该药剂的耐受性。
因此,还原剂如NAC不是很适于药团气雾剂施用。但是,预期通过肺气雾剂输注来递送还原剂将提高效力,同时允许降低吸入溶液中还原剂的浓度(预计提高耐受性)。
表面活性剂和洗涤剂是铺展剂(spreading agent),表现为降低黏液黏弹性,提高黏液清除能力。表面活性剂的实例包括二棕榈酰磷脂酰胆碱(DPPC)、PF、棕榈酸、棕榈酰-油酰磷脂酰甘油、表面活性蛋白(例如,SP-A、SP-B或SP-C)或可以是动物来源的(例如,来自母牛或小牛肺灌洗物或提取自切碎的猪肺)或其组合。参见,例如美国专利No.7,897,577、5,876,970、5,614,216、5,100,806和4,312,860。表明活性剂产品的实例包括Neonatal(棕榈胆磷(colfosceril palmitate))、(DPPC和蛋磷脂酰胆碱)、KL-4表面活性剂、(lusulptide、rSP-C表面活性剂)、(bovactant)、(poractant alfa)、(calfactant)、(修饰的牛表面活性剂)、NatsurfTM(非离子醇乙氧基化物表面活性剂)以及(beractant)。洗涤剂的实例包括但不限于Tween-80和triton-X 100。
可使用任何合适的祛痰药,包括但不限于愈创甘油醚(参见,例如美国专利No.7,345,051)。可使用任何合适的脱氧核糖核酸酶,包括但不限于Dornase Alpha.(参见,例如美国专利No.7,482,024)。激酶抑制剂的实例包括NFkB、PI3K(磷脂酰肌醇3-激酶)、p38-MAP激酶和Rho激酶的抑制剂。
用于与本发明化合物一起配制和使用的抗感染剂包括抗病毒剂和抗生素。合适的抗病毒剂的实例包括(奥司他韦)和(扎那米韦)。合适的抗生素的实例包括但不限于:氨曲南(精氨酸或赖氨酸)、磷霉素和氨基糖苷类(例如妥布霉素)或其任意组合或子集。可在本文中使用的另外抗感染剂包括氨基糖苷类、达帕托霉素、氟喹诺酮类、酮内酯类、碳青霉烯类、头孢菌素类、红霉素、利奈唑胺、青霉素类、阿奇霉素、克林霉素、唑烷酮类、四环素类和万古霉素。
可用的碳青霉烯类抗生素的实例是亚胺培南、帕尼培南、美洛培南、比阿培南、MK-826(L-749,345)、DA-1131、ER-35786、来那培南、S-4661、CS-834(R-95867的前药)、KR-21056(KR-21012的前药)、L-084(LJC11036的前药)和CXA-101。
用于与本发明化合物组合配制和使用的抗组胺剂(即,H1-受体拮抗剂)包括但不限于:乙醇胺类,例如苯海拉明HCl、马来酸卡比沙明、多西拉敏、富马酸氯马斯丁和茶苯海明(dimenhydrinate);乙二胺类,例如马来酸吡拉明(美吡拉敏)、曲吡那敏HCl、枸橼酸曲吡那敏和安他唑啉;烷基胺类,例如非尼拉敏、氯苯那敏、溴苯那敏、右氯苯那敏、曲普利啶和阿伐斯汀;吡啶类,例如美沙吡林,哌嗪类,例如羟嗪HCl、扑酸羟嗪、赛克利嗪HCl、乳酸赛克利嗪、美克洛嗪HCl和西替利嗪HCl;哌啶类,例如阿司咪唑、左卡巴斯汀HCl、氯雷他定、脱碳乙氧基氯雷他定、特非那定和非索非那定HCl;三环和四环类,例如异丙嗪、氯丙嗪、异丁嗪和阿扎他定;以及氮斯汀HCl,或者其任意组合或子集。
适合用在本文的组合和方法中的其他类的治疗剂的实例包括:抗病毒剂例如利巴韦林,抗真菌剂例如两性霉素、伊曲康唑和伏立康唑,抗排斥药物,例如环孢菌素、他克莫司和西罗莫司,支气管扩张剂,包括但不限于抗胆碱能剂例如定喘乐(atrovent)、siRNA、基因治疗载体、适配体、内皮素受体拮抗剂、α-1-抗胰蛋白酶和前列环素。
在上述处理方法和用途中,本发明化合物可单独使用或与一种或更多种其他治疗活性剂组合。通常来说,任何在本发明化合物所治疗的疾病或病症中具有治疗作用的治疗活性剂都可用于与本发明化合物组合,只要所述特定治疗活性剂与使用本发明化合物的治疗相容即可。适于与本发明化合物组合的典型治疗活性剂包括上述药剂。
在一个优选实施方案中,本发明化合物与一种或更多种渗压剂组合使用,特别是高渗盐水或甘露醇。
在另一个方面,本发明提供了上述治疗方法和用途,其包括施用有效量的本发明化合物和至少一种另外的治疗活性剂。本发明化合物和至少一种另外的治疗活性剂可以任意治疗上合适的组合伴随地或相继地组合使用。可伴随地施用本发明化合物和一种或更多种另外的治疗活性剂,这些组分存在于:1)单一药物组合物中,例如上述组合物,或2)分开的药物组合物中,其各自包含一种或更多种组分活性成分。可按照顺序方式分开施用组合的组分,其中首先施用本发明化合物,然后施用其他的治疗活性剂,反之亦然。
在其中本发明化合物与一种或更多种渗压剂组合施用的一些实施方案中,优选伴随地施用每种组分,并且可以在单个组合物中或在分开的组合物中施用。在一个实施方案中,通过经支气管镜灌洗伴随地施用本发明化合物和一种或更多种渗压剂。在另一个实施方案中,通过吸入伴随地施用本发明化合物和一种或更多种渗压剂。
当本发明化合物与另一种治疗活性剂组合使用时,每种化合物的剂量可与单独使用本发明化合物时不同。本领域的普通技术人员容易确定合适的剂量。选择本发明化合物、另外的治疗活性剂的合适的剂量和相对施用时间以实现期望的组合治疗作用,并且取决于主治医师、临床医师或兽医的专业知识和酌情处理。
实验过程如下文详细描述的,本发明还提供了用于制备本发明化合物的方法和可用在这些方法中的合成中间体。
在合成方法和实验细节的描述中使用了某些缩写和首字母缩写。但是,其大部分是本领域技术人员理解的,下表包含了许多这些缩写和首字母缩写的列表。
缩写 含义
AcOH 乙酸
AIBN 偶氮二异丁腈
DIAD 叠氮羧酸二异丙酯
DIPEA N,N-二异丙基乙胺
Cbz 羧基苄基
DCE 二氯乙烷
DCM 二氯甲烷
DMF 二甲基甲酰胺
DMSO 二甲基亚砜
Et 乙基
EtOAc或EA 乙酸乙酯
EtOH 乙醇
ESI 电喷雾离子化
HATU 2-(1H-7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐
HPLC 高效液相色谱
iCBF 氯甲酸异丁酯
iPrOH 异丙醇
i.t.或IT 气管内
Me 甲基
MeOH 甲醇
m/z或m/e 质荷比
缩写 含义
MH+ 质量加1
MH- 质量减1
MIC 最低抑制浓度
MS或ms 质谱
MTBE 甲基叔丁基醚
NaCNBH3 氰基硼氢化钠
NMM N-甲基吗啉
rt或r.t. 室温
Rf 保留因子
t-Bu 叔丁基
TEA 三乙胺
THF 四氢呋喃
TLC或tlc 薄层色谱
δ 相对于四甲基硅烷的低磁场的百万分之份数
Cbz 苄氧基羰基,即-(CO)O-苄基
AUC 曲线或峰下面积
tR 保留时间
GC-MS 气相色谱-质谱
wt% 重量百分比
h 小时
min 分钟
MHz 兆赫兹
TFA 三氟乙酸
UV 紫外线
Boc 叔丁氧羰基
缩写 含义
Ph3P 三苯基膦
可使用本领域已知的技术合成式(A)化合物。以下方案1中举例说明了代表性合成程序。
方案1
这些方法描述在例如E.J.Cragoe,“The Synthesis of Amiloride and Its Analogs”(第3章)的Amiloride and Its Analogs,第25-36页。制备阿米洛利类似物的其他方法描述在例如美国专利No.3,318,813中,特别地参见‘813专利中的方法A、B、C和D。可在本发明化合物的制备中使用的其他制备方法在PCT公开No.WO2003/07182、WO2005/108644、WO2005/022935、US 7,064,129、US 6,858,615、US 6,903,105、WO 2004/073629、WO 2007/146869和WO 2007/018640中进行描述。
通常来说,本发明的化合物可方便地通过使式(a)的化合物用式(b)的胺处理来制备。更具体地,式(a)的化合物在适当溶剂如甲醇、乙醇或四氢呋喃中加热至高温例如70℃下用式(b)的胺和碱(如三乙胺(TEA)或二异丙基乙胺(DIPEA))来处理。进一步纯化,立体异构体的分辨率、盐形式的结晶和/或制备可使用常规技术来进行。
在某些情况下,本领域的技术人员将理解,合成中起始化合物或中间化合物可具有提供交替反应位点的其他官能团。可通过利用适当的保护基如胺或醇保护基来避免干扰这样的官能团,并且在适用时适当地使合成步骤优先化。合适的保护基对于本领域技术人员而言将是显而易见的。用于设置和移除这样的保护基的方法是本领域中公知的,并且这样的常规技术也可以用于本发明的方法中。
本文中提供的以下具体实施例仅用于举例说明的目的,而不限制本发明的范围,其是由权利要求限定的。
材料和方法。所有试剂和溶剂均购自Aldrich Chemical公司、Chem-Impex International Inc.和TCI chemical industry Co.Ltd。用Bruker AC 400(1H NMR在400MHz下以及13C NMR在100MHz下)和Bruker AC 300(1H NMR在300MHz下以及13C NMR在75MHz下)获得NMR谱。质子谱参照四甲基硅烷作为内标物,碳谱参照CDCl3、CD3OD或DMSO-d6(除非另有指明,否则购自Aldrich或Cambridge Isotope Laboratories)。用装有硅胶柱(Redi Sep.Rf,Teledyne Isco)或反相柱(高性能C18Gold柱)的Combiflash系统(Combiflash Rf,Teledyne Isco)进行快速色谱。用Shimadzu LCMS-2010EV质谱仪获得ESI质谱。用Shimadzu Prominence HPLC系统使用Waters XTerra MS C18 5μm4.6×150mm分析柱在220nm(除非另有指明)检测,获得HPLC分析。流量为1.0mL/分钟,使用以下时间程序:
用Shimadzu Prominence UFLC系统使用Waters ACQUITY UPLC HSS T3 1.8μm 2.1×100mm分析柱在220nm(除非另有指明)检测,获得UPLC分析。流量为0.3mL/分钟,使用以下时间程序:
方案I.中间体1I-j和I-k的制备
3-氧代丙基氨基甲酸叔丁酯(2)的制备:
在-78℃下,向草酰氯(8.56mL、98.15mmol)的CH2Cl2(200mL)溶液中添加DMSO(8.70mL,122.5mmol)。30分钟之后,在-78℃下,添加化合物1(8.60g,49.90mmol)并且将反应混合物再搅拌30分钟。添加三乙胺(41mL,294mmol)并且在-78℃下继续将反应混合物搅拌30分钟,然后加热至0℃并且搅拌1h。反应混合物在CH2Cl2(300mL)与水(300mL)之间分配。将水层分离并用CH2Cl2(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过柱色谱(硅胶,49∶1CHCl3/MeOH)纯化,得到作为黄色液体的醛2:
1H NMR(300MHz,CD3OD)δ9.80(s,1H),4.94-4.82(br s,1H),3.42(dd,J=12.1Hz,6.0Hz,2H),2.70(t,J=6.0Hz,2H),1.43(s,9H).
(S)-4-(4-(4-(苄氧基羰基氨基)丁-1-炔基)苯基)-2-(叔丁氧基羰基氨基)丁酸(I-b)的制备:
向甲酯I-a(5.00g,10.12mmol)的THF/MeOH/H2O(60mL/60mL/20mL)溶液中添加NaOH(2.40g,60.72mmol)并且将反应混合物在室温下搅拌2h。用1N HCl水溶液将pH值调整至9并且除去有机溶剂。将残余物的pH值调整至5,并且将混悬液在CH2Cl2(500mL)与水(500mL)之间分配。将水层分离并用CH2Cl2(2×500mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩,得到作为棕色固体的化合物I-b:
1H NMR(400MHz,DMSO-d6)δ7.37-7.30(m,5H),7.27(d,J=8.4Hz,2H),7.14(d,J=8.4Hz,2H),6.73(brs,1H),5.03(s,2H),3.75-3.69(m,1H),3.21(q,J=6.4Hz,2H),2.60-2.47(m,4H),1.97-1.76(m,2H),1.38(s,9H).
(S)-4-(4-(4-(苄氧基羰基氨基)丁-1-炔基)苯基)-2-(叔丁氧基羰基氨基)丁酰胺(I-c)的制备:
在0℃下,向酸I-b(4.40g,9.10mmol)的THF(60mL)溶液中添加NMM(1.50mL,13.65mmol)和i-BCF(1.55mL,11.91mmol)。反应混合物在相同温度下搅拌2h并且逐滴添加NH3(甲醇中的7.0N,13mL,91mmol)。反应混合物继续在0℃下搅拌2h,然后加热至室温并搅拌16h。浓缩之后,残余物在CH2Cl2(300mL)与水(300mL)之间分配。将水层分离并用CH2Cl2(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过柱色谱(硅胶,30∶1CHCl3/MeOH)纯化,得到作为黄色固体的酰胺I-c:
1H NMR(400MHz,CD3OD)δ7.36-7.24(m,7H),7.13(d,J=8.4Hz,2H),5.08(s,2H),4.02-3.96(m,1H),2.72-2.56(m,2H),2.57(t,J=7.1Hz,2H),2.07-1.86(m,4H),1.45(s,9H).
化合物(I-d)的制备:
在室温下使I-c(3.40g,7.0mmol)和10%林德拉催化剂(2.00g)的EtOH(100mL)混悬液处于氢化条件(1atm)36h。该反应混合物经硅藻土过滤并用MeOH洗涤。在真空下浓缩滤液并将残余物通过柱色谱(硅胶,95∶5CHCl3/CH3OH)纯化,得到作为黄色固体的化合物I-d:
1H NMR(300MHz,CD3OD)δ7.35-7.23(m,5H),7.21-7.12(m,2H),7.07(s,2H),5.04(s,2H),4.06-3.94(m,1H),3.21(t,J=7.0Hz,1H),3.11(t,J=7.0Hz,1H),2.77-2.46(m,4H),2.10-1.77(m,4H),1.67-1.51(m,2H),1.45(s,9H).
化合物(I-e)的制备:
在室温下,将化合物I-d(2.9g,6.0mmol)溶解于二氧六环(20mL)中的4N HCl中并将该溶液搅拌1h。在真空下除去溶剂,得到作为白色固体的化合物I-e:
1H NMR(300MHz,CD3OD)δ7.34-7.23(m,5H),7.22-7.14(m,2H),7.11(s,2H),5.05(s,2H),3.98-3.90(m,1H),3.21(t,J=7.0Hz,1H),3.11(t,J=7.0Hz,1H),2.69(dd,J=17Hz,8.0Hz,2H),2.57(t,J=6.9Hz,1H),2.50(ddd,J=9.3Hz,7.5Hz,2.1Hz,1H),2.00-2.04(m,3H),1.67-1.45(m,3H).
化合物(I-f)的制备:
向化合物I-e(2.40g,5.72mmol)和醛2(1.2g,6.87mmol)的MeOH(35mL)溶液中添加乙酸(0.5mL)并在室温下将反应混合物搅拌10分钟。然后添加氰基硼氢化钠(540mg,8.58mmol)并将该溶液在室温下继续搅拌3h。添加额外的化合物2(0.3当量)、AcOH(0.5当量)和NaCNBH3(0.5当量)并将该溶液在室温下继续搅拌12h。浓缩之后,残余物在EtOAc(300mL)和饱和NaHCO3(300mL)之间分配。将水层分离并用EtOAc(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物(粗制I-f,3.50g)无需进一步纯化直接用于下一个步骤。化合物(I-g)的制备
在0℃下,向化合物I-f(粗制,3.50g)的MeOH(25mL)溶液添加饱和NaHCO3(25mL)并且将该溶液搅拌10分钟。逐滴添加氯甲酸苄酯(1.75mL)并将该反应混合物在0℃下搅拌2h,然后加热至室温并搅拌1h。浓缩之后,将残余物溶解在CH2Cl2(200mL)中,然后用水(300mL)和盐水(300mL)洗涤。有机层经Na2SO4干燥并浓缩。残余物(粗制I-g,3.50g)无需进一步纯化直接用于下一个步骤。
化合物(I-h)的制备:
在室温下,将化合物I-g(粗制,3.50g)溶解在二氧六环中的4N HCl(30mL)中并将该溶液搅拌1h。浓缩之后,残余物用NH4OH水溶液中和并通过柱色谱(硅胶,16∶1CHCl3/MeOH)纯化,得到作为黄色固体的化合物I-h:
1H NMR(300MHz,CD3OD)7.45-7.22(m,10H),7.21-6.99(m,4H),5.15(s,2H),5.04(s,2H),4.54-4.36(m,1H),3.55-3.39(m,2H),3.21(t,J=7.2Hz,1H),3.11(t,J=7.2Hz,1H),2.97-2.80(m,2H),2.56(t,J=7.2Hz,2H),2.52-2.44(m,4H),2.28-2.01(m,2H),1.91(t,J=6.6Hz,2H),1.64-1.43(m,2H).
化合物I-j和化合物I-k的制备
向化合物I-h(1.15g,2.00mmol)和三醇Ii(2.68g,10.0mmol)的甲醇(35mL)溶液添加乙酸(0.91mL)并且将反应混合物在室温下搅拌10分钟。添加氰基硼氢化钠(880mg,14.0mmol)并将该溶液在室温下继续搅拌2h。添加额外的化合物I-i(6.0当量)、AcOH(8.0当量)和NaCNBH3(8.0当量)并将该溶液在室温下继续搅拌16h。添加己醛(0.36mL,3.00mmol)、AcOH(0.91mL)和NaCNBH3(0880mg,14.0mmol)并将反应混合物搅拌2h。浓缩之后,残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。将水层分离并用EtOAc(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过C-18反相Gold柱来纯化,得到作为白色固体的化合物I-j和化合物I-k。
2-((1-氨基-4-(4-(4-(苄氧基羰基氨基)丁基)苯基)-1-氧代丁烷-2-基)(3-(((2S,3R)-2,3-二羟基-3-((2S,4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)氨基)丙基)氨基)乙酸苄酯(化合物I-j)的数据:
1H NMR(400MHz,CD3OD)δ7.53-7.39(m,4H),7.37-7.22(m,16H),7.18-6.94(m,4H),5.52-5.37(m,2H),5.09(s,2H),5.04(s,2H),4.21(dd,J=11Hz,5.5Hz,2H)4.01-3.89(m,4H),3.88-3.81(m,2H),3.75-3.64(m,3H),3.57(t,J=9.5Hz,2H),3.19(t,J=7.1Hz,1H),3.09(t,J=6.4Hz,2H),2.75-2.40(m,12H),2.25-2.08(m,1H),2.04-1.86(m,1H),1.85-1.65(m,3H),1.63-1.53(m,1H),1.52-1.40(m,1H).
2-((1-氨基-4-(4-(4-(苄氧基羰基氨基)丁基)苯基)-1-氧代丁烷-2-基)(3-(((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)(己基)氨基)丙基)氨基)乙酸苄酯(化合物I-k)的数据:
1H NMR(400MHz,CD3OD)of I-k,δ7.58-7.22(m,15H),7.20-6.95(m,4H),5.53-5.43(m,1H),5.11(s,2H),5.05(s,2H),4.22(dd,J=9.7Hz,4.8Hz,1H)4.00-3.83(m,4H),3.80-3.69(m,1H),3.59(t,J=10.6Hz,1H),3.20(t,J=6.8Hz,1H),3.10(t,J=5.8Hz,2H),2.77-2.62(m,1H),2.61-2.31(m,10H),2.27-2.14(m,1H),2.06-1.89(m,1H),1.86-1.54(m,3H),1.53-1.44(m,1H),1.41-1.00(m,10H),0.85(t,J=5.9Hz,3H).
方案II.3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(II-d)的盐酸盐的制备
化合物II-a的制备:
在室温下,使I-j(700mg,0.65)和10%Pd/C(300mg)的EtOH/AcOH(40mL/4mL)混悬液处于氢化条件(1atm)下16h。反应混合物通过硅藻土过滤并用MeOH洗涤。在真空下浓缩滤液,得到作为白色固体的化合物II-a:
1H NMR(400MHz,CD3OD)δ7.47-7.40(m,4H),7.35-7.26(m,7H),7.10-7.07(m,3H),5.49(s,2H),4.24(dd,J=10.7Hz,5.4Hz,1H),4.18-4.09(m,2H),4.00-3.88(m,3H),3.87-3.82(m,2H),3.77-3.69(m,2H),3.59(t,J=10.0Hz,2H),3.54-3.46(m,1H),3.06(dd,J=12.7Hz,J=9.0Hz,1H),3.00-2.93(m,1H),2.92(t,J=8.1Hz,2H),2.83-2.71(m,4H),2.66-2.51(m,4H),2.06-1.85(m,4H),1.95(s,9H),1.73-1.57(m,4H),1.38-1.00(m,2H).
化合物II-c的制备
在室温下,向化合物II-a(650mg,0.65mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯(methyl3,5-diamino-6-chloropyrazine-2-carbonylcarbamimidothioate)氢碘酸盐(II-b,409mg,1.05mmol)的EtOH(25mL)溶液中添加DIPEA(0.92mL,5.20mmol)。在密封管中于70℃下将反应混合物加热2h,然后冷却至室温并在真空下浓缩。残余物通过柱色谱(硅胶,9∶1CH2Cl2/MeOH,80∶18∶2CHCl3/CH3OH/NH4OH)纯化,得到作为黄色固体的化合物II-c:
1H NMR(400MHz,CD3OD)δ7.48-7.41(m,4H),7.35-7.26(m,6H),7.10(brs,4H),5.45(s,2H),4.22(dd,J=10.7Hz,5.2Hz,2H),4.00-3.90(m,4H),4.00-3.88(m,2H),3.85(dd,J=5.4Hz,3.0Hz,2H),3.71(t,J=2.3Hz,1H),3.69(t,J=2.3Hz,1H),3.58(t,J=11.4Hz,2H),3.25(t,J=7.6Hz,2H),3.05(t,J=6.7Hz,1H),2.73-2.41(m,6H),1.89-1.77(m,2H),1.75-1.53(m,6H).
3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(II-d)的盐酸盐的制备:
在室温下将化合物II-c(260mg,0.25mmol)的1N HCl(25mL)水溶液搅拌2h。除去溶剂并将残余物通过C-18反相Gold柱来纯化,得到作为黄色吸湿性固体的化合物II-d:
1H NMR(400MHz,CD3OD)δ7.23-7.12(m,4H),4.29-4.18(m,2H),4.05(t,J=6.2Hz,1H),3.90-3.84(m,2H),3.82-3.79(m,1H),3.78-3.76(m,1H),3.74-3.63(m,6H),3.61-3.40(m,8H),3.34(t,J=6.8Hz,2H),3.26-3.08(m,2H),2.76-2.61(m,4H),2.36-2.25(m,2H),2.24-2.15(m,2H),1.81-1.67(m,4H).
方案III.3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(III-d)的盐酸盐的制备:
III-a-(2S)-4-(4-(4-氨基丁基)苯基)-2-(3-(((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)(己基)氨基)丙基氨基)丁酰胺三乙酸盐的制备
在室温下,使I-k(450mg,0.54)和10%Pd/C(200mg)的EtOH/AcOH(20mL/2mL)混悬液处于氢化条件(1atm)6h。反应混合物通过硅藻土过滤并用MeOH洗涤。在真空下浓缩滤液,得到作为白色固体的化合物III-a:
1H NMR(400MHz,CD3OD)δ7.50-7.43(m,2H),7.36-7.30(m,3H),7.13-7.09(m,4H),5.54(s,1H),4.25(dd,J=10.7Hz,5.1Hz,2H),4.17(ddd,J=9.3Hz,5.8Hz,3.1Hz,1H),3.98(dd,J=9.5Hz,5.3Hz,1H),3.91(dd,J=5.6Hz,1.8Hz,1H),3.78(dd,J=9.5Hz,2.2Hz,1H),3.62(t,J=10.4Hz,1H),3.29-3.16(m,2H),3.14-3.0(m,2H),3.04-2.94(m,1H),2.90(t,J=7.1Hz,2H),2.72(t,J=5.3Hz,2H),2.67-2.57(m,4H),1.94(s,9H),1.91-1.84(m,2H),1.83-1.76(m,2H),1.73-.1.54(m,6H),1.33-1.15(m,7H),0.86(t,J=8.1Hz,3H)
III-c-3,5-二氨基-N-(N-(4-(4-((3S)-4-氨基-3-(3-(((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)(己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的制备
在室温下,向化合物III-a(400mg,0.48mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(III-b,302mg,0.77mmol)的EtOH(15mL)溶液中添加DIPEA(0.68mL,3.84mmol)。在密封管中于70℃下将反应混合物加热2h,然后冷却至室温并在真空中浓缩。残余物通过柱色谱(硅胶,9∶1CH2Cl2/MeOH,80∶18∶2CHCl3/CH3OH/NH4OH)纯化,得到作为黄色固体的化合物III-c:
1H NMR(400MHz,CD3OD)δ7.46(dd,J=8.0Hz,4.6Hz,2H),7.33-7.28(m,3H),7.11(brs,4H),5.52(s,1H),4.23(dd,J=10.8Hz,5.2Hz,1H),4.02-3.92(m,2H),3.89(dd,J=5.3Hz,2.1Hz,1H),3.75(dd,J=9.3Hz,2.2Hz,1H),3.60(t,J=10.5Hz,1H),3.29-3.21(m,2H),3.07(t,J=7.1Hz,1H),2.76(dd,J=12.7Hz,5.6Hz,2H),2.68-2.41(m,10H),1.93-1.78(m,2H),1.76-.1.53(m,6H),1.47-1.37(m,2H),1.32-1.16(m,6H),0.86(t,J=8.1Hz,3H).
3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(III-d)的盐酸盐的制备:
在室温下,将化合物III-c(230mg,0.27mmol)的1NHCl(20mL)水溶液搅拌3h。除去溶剂并将残余物通过C-18反相Gold柱来纯化,得到作为黄色吸湿性固体的化合物III-d:
1H NMR(400MHz,CD3OD)δ7.16(s,4H),4.18(dd,J=12.0Hz,5.6Hz,1H),4.03-3.95(m,1H),3.84(dd,J=4.9Hz,1.2Hz,1H),3.78(dd,J=10.5Hz,3.0Hz,1H),3.74-3.63(m,3H),3.42-3.32(m,6H),3.28-3.21(m,2H),3.19-3.04(m,2H),2.76-2.62(m,4H),2.30-2.13(m,4H),1.84-1.65(m,6H),1.46-1.32(m,6H),0.93(t,J=7.2Hz,3H).
方案IV.中间体IV-x和中间体IV-y的制备
化合物IV-d-4-(4-甲氧基萘-1-基)丁酸的制备
在室温下,向4-(4-甲氧基萘-1-基)-4-氧代丁酸、化合物IV-c(100g,387.5mmol)的甲苯(500mL)和浓盐酸(500mL)溶液中分批加入Zn粉(251g、3.87mol)。将反应混合物回流加热2h,冷却至室温并通过硅藻土过滤。在将滤液浓缩至50%之后,过滤所得的沉淀并干燥,得到作为灰白色(off-white)固体的化合物IV-d:
1H NMR(400MHz,DMSO-d6)δ12.05(s,1H),8.18(dd,J=8.0Hz,1.2Hz,1H),8.04(d,J=8.0Hz,1H),7.58-7.47(m,2H),7.26(d,J=7.6Hz,1H),6.89(d,J=8.0Hz,1H),3.94(s,3H),2.96(t,J=7.6Hz,2H),2.30(t,J=7.6Hz,2H),1.85(t,J=7.6Hz,2H).
化合物IV-e-(S)-4-苄基-3-(4-(4-甲氧基萘-1-基)丁酰基)唑烷-2-酮的制备
在-78℃下,向化合物IV-d(43.5g,245.9mmol)的无水THF(500mL)溶液中逐滴添加正丁基锂并且将反应混合物搅拌45分钟,得到化合物IV-b的溶液。在0℃,向化合物IV-d(50.0g,204.9mmol)的无水THF(100mL)独立溶液中逐滴加入NMM(25.0g,245.9mmol)和i-BCF(30.7g,245.9mmol)。在相同的温度下,将反应混合物再搅拌30分钟,并且然后在0℃下缓慢地添加化合物的制备溶液。将反应混合物在室温下再搅拌3h,用饱和NH4Cl淬灭,浓缩以除去THF,并且在EtOAc(1000mL)与水(1000mL)之间分配。将水层分离并用EtOAc(2×1000mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过柱色谱(硅胶,30∶70EtOAc/己烷)纯化,得到作为白色固体的化合物IV-e:
1H NMR(400MHz,DMSO-d6)δ8.19(d,J=8.3,1H),8.09(d,J=8.3,1H),7.60-7.48(m,2H),7.31-7.19(m,6H),6.91(d,J=7.8Hz,1H),4.69-4.63(m,1H),4.31(t,J=8.6Hz,1H),4.16(dd,J=8.8Hz,2.8Hz,1H),3.95(s,3H),3.03-2.99(m,2H),2.95-2.89(m,2H),1.98-1.93(m,2H).
化合物IV-f-(S)-3-((S)-2-叠氮基-4-(4-甲氧基萘-1-基)丁酰基)-4-苄基唑烷-2-酮的制备
在78℃下,向化合物IV-e(10.0g,24.81mmol)的无水THF(70mL)溶液逐份加入KHMDS(6.40g,32.3mmol)。将所得的混合物搅拌30分钟之后,添加三异丙基苯磺酰叠氮化物(trisyl azide,11.5g,37.2mmol)并将反应混合物搅拌2分钟至3分钟。在相同温度下缓慢添加乙酸(9.0g,148.8mmol)随后添加四甲基乙酸铵(13.2g,99.24mmol)。将反应混合物加热至27℃,搅拌16h,用饱和NaHCO3(300mL)淬灭,浓缩以除去THF并且用EtOAc(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过柱色谱(硅胶,30∶70EtOAc/己烷)纯化,得到作为无色油状物的化合物IV-f:
1H NMR(300MHz,CDCl3)δ8.30(t,J=6.1Hz,1H),7.99(d,J=8.3Hz,1H),7.56-7.47(m,2H),7.37-7.28(m,3H),7.20-7.16(m,2H),6.74(d,J=7.8Hz,1H),5.12-5.08(m,1H),4.54-4.49(m,1H),4.16-4.11(m,2H),3.98(s,3H),3.33-3.27(m,3H),2.84-2.77(m,1H),2.35-2.04(m,2H).
化合物IV-g-(S)-2-叠氮基-4-(4-甲氧基萘-1-基)丁酸的制备
在0℃,向化合物IV-f(6.10g,13.7mmol)的THF/H2O(70mL/30mL)溶液添加H2O2(2.80g,82.2mmol)随后分批添加LiOH(1.15g,27.4mmol)。在相同温度下,将反应混合物搅拌3h,用饱和Na2SO3(200mL)淬灭,在减压下浓缩以除去THF并用CH2Cl2(200mL)洗涤。水层用1N HCl水溶液酸化并且用CH2Cl2(2×250mL)萃取。合并的有机萃取物经Na2SO4干燥,浓缩并用MTBE洗涤,得到作为灰白色固体的化合物IV-g:
1H NMR(400MHz,DMSO-d6)δ13.15(s,1H),8.19(dd,J=8.0Hz,1.2Hz,1H),8.00(d,J=8.4Hz,1H),7.60-7.48(m,2H),7.29(d,J=7.8Hz,1H),6.90(d,J=7.8Hz,1H),4.21-4.18(m,1H),3.94(s,3H),3.07-3.04(m,2H),2.13-1.89(m,2H).
化合物IV-h-(S)-2-氨基-4-(4-甲氧基萘-1-基)丁酸乙酯的制备
在室温下,使化合物IV-g(3.20g,11.2mmol)和10%Pd/C(1.60g)的AcOH/H2O(80mL/20mL)混悬液处于氢化条件(1atm)3h。反应混合物通过硅藻土过滤并用MeOH洗涤。在真空下浓缩滤液,得到作为黄色固体的乙酸盐IV-h:
1H NMR(300MHz,DMSO-d6)δ13.15(s,1H),8.18(d,J=8.1Hz,1H),8.08(d,J=8.1Hz,1H),7.57-7.46(m,2H),7.28(d,J=7.6Hz,1H),6.89(d,J=7.8Hz,1H),3.93(s,3H),3.04(t,J=6.9Hz,2H),2.08-1.90(m,2H).
化合物IV-i-(S)-2-氨基-4-(4-羟基萘-1-基)丁酸氢溴酸盐的制备
在室温下,向化合物IV-h(2.80g,10.81mmol)的乙酸(30mL)溶液逐滴添加氢溴酸(30mL)并且将反应混合物回流4h。将反应混合物冷却至室温并浓缩。残余物用H2O(15mL)稀释,用氨水稍微碱化并过夜结晶,得到作为棕色固体的化合物IV-i:ESI-MS m/z 246[C14H15NO3+H]+。化合物IV-j-(S)-2-氨基-4-(4-羟基萘-1-基)丁酸甲酯的制备
在0℃下,将乙酰氯(13.5g,171.4mmol)加入无水甲醇(70mL)并且随后添加化合物IV-i(6.00g,24.48mmol)。将反应混合物回流4h并浓缩。残余物在CH2Cl2(300mL)与饱和NaHCO3(300mL)之间分配。将水层分离并用CH2Cl2(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩,得到作为无色油状物的化合物IV-j:ESI-MS m/z 260[C15H17NO3+H]+。
化合物IV-k-(S)-2-(叔丁氧基羰基氨基)-4-(4-羟基萘-1-基)丁酸甲酯的制备
在0℃下,向化合物IV-j(4.80g,18.53mmol)的MeOH/H2O(40mL/10mL)溶液添加NaHCO3(6.20g,74.13mmol)和Boc2O(4.85g,22.2mmol)。将所得的混合物加热至室温并搅拌3h。反应混合物在CH2Cl2(200mL)与水(200mL)之间分配。将水层分离并用CH2Cl2(2×200mL)萃取。合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为无色油状物的化合物IV-k:
1H NMR(300MHz,DMSO-d6)δ9.91(s,1H),8.15(dd,J=8.0Hz,1.2Hz,1H),7.98(d,J=8.3Hz,1H),7.52-7.41(m,3H),7.13(d,J=7.7Hz,1H),6.77(d,J=7.6Hz,1H),4.05-3.99(m,1H),3.66(s,3H),3.04-2.86(m,2H),1.97-1.91(m,2H),1.42(s,9H).
化合物IV-l-(S)-2-(叔丁氧基羰基氨基)-4-(4-(三氟甲基磺酰氧基)萘-1-基)丁酸甲酯的制备
在0℃下,向化合物IV-k(9.50g,26.46mmol)的吡啶(50mL)溶液中添加三氟甲磺酸酐(8.90g,282.1mmol),并且将反应混合物在室温下搅拌2.5h。浓缩之后,反应混合物在CH2Cl2(300mL)与水(300mL)之间分配。将水层分离并用CH2Cl2(2×300mL)萃取。合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为棕色油状物的化合物IV-l:ESI-MS m/z 492[C21H24F3NO7S+H]+。
化合物IV-n-(S)-4-(4-(4-(苄氧基羰基氨基)丁-1-炔基)萘-1-基)-2-(叔丁氧基羰基氨基)丁酸甲酯的制备
在室温下,向化合物IV-l(6.00g,12.21mmol)的无水CH3CN(100mL)溶液中添加TEA(4.93g,48.8mmol)、己烷中的10%(t-Bu)3P(0.50g,2.44mmol)、丁-3-炔基氨基甲酸苄酯(IV-m,3.70g,18.3mmol)和CuI(0.11g,0.61mmol)。所得的混合物用氩气脱气3分钟并一次性迅速添加Pd(PPh3)4(1.40g,1.22mmol)。用氩气脱气5分钟之后,将所得的混合物回流5h。真空中浓缩反应混合物并将残余物通过柱(硅胶,40∶60己烷/EA)来纯化,得到作为棕色油状物的化合物IV-n:
1H NMR(300MHz,CDCl3)δ8.35-8.31(m,1H),7.97-7.94(m,1H),7.54-7.50(m,3H),7.37-7.29(m,6H),5.29-5.13(m,4H),4.44(br s,1H),3.73(s,3H),3.56-3.49(m,2H),3.14-3.07(m,2H),2.79(t,J=6.6Hz,2H),2.10-1.98(m,2H),1.42(s,9H).
化合物IV-o-(S)-4-(4-(4-(苄氧基羰基氨基)丁-1-炔基)萘-1-基)-2-(叔丁氧基羰基氨基)丁酸的制备
向甲酯IV-n(3.40g,6.25mmol)的THF/MeOH/H2O(30mL/30mL/10mL)溶液中添加NaOH(0.75g,18.7mmol)并且将反应混合物在室温下搅拌3h。用1N HCl水溶液将pH值调整至9并且除去有机溶剂。将pH值调整到5,并且该混悬液在CH2Cl2(200mL)与水(200mL)之间分配。将水层分离并用CH2Cl2(2×200mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩,得到作为棕色固体的化合物IV-o:
1H NMR(300MHz,CDCl3)δ8.28(t,J=7.0Hz,1H),7.91(br s,1H),7.45(d,J=6.9Hz,3H),7.34-7.29(m,6H),7.15(d,J=6.3Hz,1H),5.29-5.12(m,4H),4.31(br s,1H),3.51(d,J=6.2Hz,2H),3.06(br s,1H),2.76(t,J=6.2Hz,2H),2.30-2.04(m,2H),1.42(s,9H).
化合物IV-p-(S)-4-(4-(4-(苄氧基羰基氨基)丁-1-炔基)萘-1-基)-2-(叔丁氧基羰基氨基)丁酸的制备
在0℃下,向酸IV-o(2.90g,5.47mmol)的THF(40mL)溶液添加NMM(0.82g,8.2mmol)和i-BCF(0.97g,7.11mmol)。将反应混合物在相同温度下搅拌30分钟并且逐滴添加NH3(7.0N溶于甲醇,6.0mL,43.7mmol)。反应混合物在0℃下继续搅拌2h,加热至室温并搅拌16h。浓缩之后,残余物在CH2Cl2(100mL)与水(100mL)之间分配。将水层分离并用CH2Cl2(2×100mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物用MTBE洗涤,得到作为黄色固体的酰胺IV-p:
1H NMR(400MHz,DMSO-d6)δ8.29(br s,1H),8.13(br s,1H),7.59-7.53(m,3H),7.34-7.28(m,9H),7.03(t,J=7.2Hz,1H),5.05(s,2H),3.98(br s,1H),3.31(br s,2H),3.13-3.01(m,3H),2.72(t,J=6.6Hz,2H),1.98-1.84(m,2H),1.42(s,9H).
化合物IV-q-(S)-1-氨基-4-(4-(4-氨基丁基)萘-1-基)-1-氧代丁烷-2-基氨基甲酸叔丁酯的制备
在室温下,使化合物IV-p(2.30g,4.34mmol)和10%Pd/C(1.20g)的EtOH(50mL)混悬液处于氢化条件(1atm)16h。反应混合物通过硅藻土过滤并用EtOH洗涤。在真空中浓缩滤液并用MTBE/己烷洗涤,得到作为灰白色固体的乙酸盐IV-q:
1H NMR(400MHz,CDCl3)δ8.05-8.01(m,2H),7.50-7.42(m,2H),7.24-7.20(m,2H),6.22(br s,1H),5.50(br s,1H),5.16(d,J=8.0Hz,1H),4.20(br s,1H),3.12(t,J=8.1Hz,2H),3.04(t,J=7.5Hz,2H),2.71(t,J=7.0Hz,2H),2.34-2.24(m,1H),2.07-1.98(m,1H),1.80-1.70(m,6H),1.45(s,9H).
化合物IV-r的制备
在0℃下向化合物IV-q(1.4g,3.50mmol)的干燥CH2Cl2(25mL)的搅拌溶液添加TEA(0.53g,5.25mmol)和CbzCl(0.65g,3.85mmol)。将反应混合物在室温下搅拌2h并在CH2Cl2(100mL)与水(100mL)之间分配。将水层分离并用CH2Cl2(2×100mL)萃取。合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为黄色油状物的化合物IV-r:ESI-MS m/z 534[C31H39N3O5+H]+。
化合物IV-s的制备
向化合物IV-r(1.75g,3.28mmol)的无水THF(10mL)的溶液中添加二氧六环中的4N HCl(20mL)并且将反应混合物在室温下搅拌6h。真空中除去溶剂并用MTBE洗涤残余物,得到作为灰白色固体的化合物IV-s:
1H NMR(400MHz,MeOD-d3)δ8.07-8.03(m,2H),7.53-7.49(m,2H),7.31-7.27(m,7H),5.05(s,2H),4.05(t,J=6.2Hz,1H),3.20-3.14(m,6H),2.26-2.22(m,2H),1.76-1.59(m,4H).
化合物IV-t的制备
向化合物IV-s(1.2g,2.77mmol)和醛2(0.95g,5.54mmol)的MeOH(25mL)溶液添加乙酸(0.5mL)并且将反应混合物在室温下搅拌10分钟。添加氰基硼氢化钠(0.25g,4.15mmol)并将该溶液在室温下继续搅拌16h。在3h的时间中添加额外的化合物2(0.3当量)、AcOH(0.3当量)和NaCNBH3(0.3当量)并且重复这种添加四次直到LC-MS显示出胺的消耗>90%。浓缩之后,残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。将水层分离并用EtOAc(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩,得到IV-t,其无需进一步纯化即用于下一个步骤。
化合物IV-u的制备
在0℃下,向化合物IV-t(粗制,850mg)的MeOH/H2O(25mL/12mL)溶液添加NaHCO3(0.36g,4.32mmol)并且将该溶液搅拌10分钟。逐滴添加氯甲酸苄酯(0.50g,2.88mmol)并将反应混合物在0℃下搅拌2h,然后加热至室温并搅拌1h。浓缩之后,残余物溶解在CH2Cl2(200mL)中,然后用水(300mL)和盐水(300mL)洗涤。有机层经Na2SO4干燥并浓缩,得到IV-u,其无需进一步纯化即用于下一个步骤。
化合物IV-v的制备
在室温下,将化合物IV-u(粗制,750mg)溶于二氧六环中的4N HCl(10mL)中并将该溶液搅拌2h。浓缩之后,残余物用MTBE洗涤并用NH4OH水溶液中和,得到作为灰白色固体的化合物IV-v:
1H NMR(400MHz,CD3OD)δ8.06(br s,2H),7.48-7.46(m,2H),7.31-7.19(m,11H),5.48(s,2H),5.13-5.04(m,4H),3.34-3.32(m,3H),3.15(t,J=6.9Hz,4H),3.04(t,J=7.5Hz,2H),2.58-2.40(m,3H),2.12(br s,1H),1.76-1.57(m,6H).
化合物IV-x和化合物IV-y的制备
向化合物IV-v(710mg,1.13mmol)和三醇IV-w(1.52g,5.68mmol)的甲醇(50mL)溶液添加乙酸(1.00mL)并将反应混合物在室温下搅拌30分钟。添加氰基硼氢化钠(490mg,7.91mmol)并将该溶液在室温下继续搅拌16h。添加额外的化合物IV-w(16.0当量)、AcOH(20.0当量)和NaCNBH3(20.0当量)并且将该溶液在室温下继续搅拌72h。添加己醛(0.45g,4.52mmol)、AcOH(0.91mL)和NaCNBH3(350mg,5.65mmol)并将反应混合物搅拌4h。浓缩之后,残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。将水层分离并用EtOAc(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过C-18反相Gold柱纯化,得到作为白色固体的化合物IV-x和化合物IV-y:
2-(((S)-1-氨基-4-(4-(4-(苄氧基羰基氨基)丁基)萘-1-基)-1-氧代丁烷-2-基)(3-(双((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)氨基)丙基)氨基)乙酸苄酯(化合物IV-x)的数据:
ESI-MS m/z 1130[C63H76N4O15+H]+。
2-(((S)-1-氨基-4-(4-(4-(苄氧基羰基氨基)丁基)萘-1-基)-1-氧代丁烷-2-基)(3-(((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)(己基)氨基)丙基)氨基)乙酸苄酯(化合物IV-y)的数据:
ESI-MS m/z 962[C56H72N4O10+H]+。
方案V.3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺盐酸盐的制备
V-a-(2S)-4-(4-(4-氨基丁基)萘-1-基)-2-(3-(双((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)氨基)丙基氨基)丁酰胺乙酸盐的制备
在室温下,使IV-x(425mg,0.38mmol)和10%Pd/C(200mg)的MeOH/AcOH(5.0mL/1.0mL)混悬液处于氢化条件(1atm)8h。反应混合物通过硅藻土来过滤并用MeOH洗涤。在真空中浓缩滤液并从MTBE/己烷中沉淀,得到作为无色油状物的化合物V-a:
1H NMR(300MHz,CD3OD)δ8.10-8.05(m,2H),7.55-7.21(m,14H),5.40(s,2H),4.24-4.20(m,2H),3.94-3.92(m,6H),3.86-3.53(m,12H),3.15-3.01(m,8H),2.92-2.75(m,4H),1.94(s,9H),1.76-1.33(m,8H)
V-c-3,5-二氨基-N-(N-(4-(4-((3S)-4-氨基-3-(3-(双((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的制备
在室温下,向化合物V-a(356mg,0.34mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(V-b,213mg,0.54mmol)的EtOH(8.0mL)溶液中添加DIPEA(353mg,2.73mmol)。在密封管中70℃下将反应混合物加热2h,然后冷却至室温并在真空中浓缩。残余物通过柱色谱(硅胶,10∶1CH2Cl2/MeOH,8∶2∶0.2CHCl3/CH3OH/NH4OH)纯化,得到作为黄色固体的化合物V-c:ESI-MS m/z 537[C53H69CIN10O12+2H]2+/2。
V-d-3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺盐酸盐的制备
在室温下将化合物V-c(120mg,0.111mmol)的1N HCl(3.0mL)水溶液和MeOH中的3N HCl(3.0mL)的溶液搅拌3h。除去溶剂并通过C-18反相Gold柱纯化残余物,得到作为黄色吸湿性固体的化合物V-d:
1H NMR(400MHz,D2O)δ8.13(d,J=8.4Hz,1H),7.82(d,J=9.2Hz,1H),7.53-7.31(m,4H),4.18-4.16(m,2H),3.95(brs,1H),3.78-3.74(m,6H),3.62-3.58(m,4H),3.36-3.29(m,6H),3.22-3.18(m,2H),3.08-3.04(m,6H),2.17-2.14(m,4H),1.85-1.73(m,4H);ESI-MS m/z 449[C39H61CIN10O12+2H]2+/2.
方案VI.3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(VI-d)的盐酸盐的制备
VI-a-(2S)-4-(4-(4-氨基丁基)萘-1-基)-2-(3-(((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)(己基)氨基)丙基氨基)丁酰胺三乙酸盐的制备
在室温下,使IV-y(270mg,0.28mmol)和10%Pd/C(120mg)的MeOH/AcOH(5.0mL/1.0mL)混悬液处于氢化条件(1atm)8h。反应混合物通过硅藻土来过滤并用MeOH洗涤。真空中浓缩滤液,得到无色油状物的化合物VI-a:
1H NMR(300MHz,CD3OD)δ8.09-8.05(m,2H),7.54-7.21(m,9H),5.46(s,1H),4.24-4.21(m,2H),3.95-3.94(m,2H),3.79-3.59(m,5H),3.15-3.07(m,6H),2.96-2.89(m,2H),2.76-2.75(m,2H),1.94(s,9H),1.90-1.62(m,10H),1.28-1.22(m,6H),0.78-0.76(m,3H)
化合物VI-c-3,5-二氨基-N-(N-(4-(4-((3S)-4-氨基-3-(3-(((2S,3R)-2,3-二羟基-3-((4R,5R)-5-羟基-2-苯基-1,3-二烷-4-基)丙基)(己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的制备
在室温下,向化合物VI-a(209mg,0.24mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(VI-b,130mg,0.38mmol)的EtOH(5.0mL)溶液添加DIPEA(250mg,1.92mmol)。在密封管中70℃下将反应混合物加热2h,然后冷却至室温并在真空中浓缩。残余物通过柱色谱(硅胶,10∶1CH2Cl2/MeOH,8∶2∶0.2CHCl3/CH3OH/NH4OH)纯化,得到作为黄色固体的化合物VI-c:
1H NMR(400MHz,CD3OD)δ8.08-8.06(m,2H),7.50-7.41(m,4H),7.28-7.26(m,5H),5.48(s,1H),4.23(dd,J=10.4Hz,5.2Hz,1H),3.90-3.88(m,3H),3.75(dd,J=9.6Hz,2.4Hz,1H),3.58(t,J=10.4Hz,1H),3.20-3.12(m,5H),2.70-2.48(m,7H),1.88-1.62(m,8H),1.43-1.41(m,2H),1.26-1.17(m,6H),0.83(t,J=8.1Hz,3H)
化合物VI-d-3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺盐酸盐的制备
将化合物VI-c(98mg,0.108mmol)的1N HCl(2.0mL)水溶液和MeOH中的3N HCl(2.0mL)的溶液在室温下搅拌2h。除去溶剂并通过C-18反相Gold柱纯化残余物,得到作为黄色吸湿性固体的化合物VI-d:
1H NMR(400MHz,D2O)δ8.12(d,J=8.8Hz,1H),7.83(d,J=8.0Hz,1H),7.51-7.30(m,4H),4.14-4.12(m,1H),3.76-3.58(m,7H),3.39-2.90(m,14H),2.17-1.62(m,10H),1.28-1.19(m,6H),0.78(t,J=6.4Hz,3H);ESI-MS m/z409[C39H61CIN10O7+2H]2+/2.
方案VII.3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(化合物VII-ee)的制备:
化合物VII-c的制备
将300mL无水CH2Cl2中的膦酰乙酸三甲酯VII-b(34.8mL,241mmol)冷却至0℃并且加入DBU(30.5mL,322mmol),并且将该混合物搅拌15分钟。逐滴加入50mL CH2Cl2中的醛VII-a(25.0g,134mmol)。将反应混合物恢复至室温,搅拌36h并用100mL的水淬灭。分配混合物并且将水层用CH2Cl2(3×150mL)萃取。合并的有机层用盐水洗涤,干燥(Na2SO4),过滤并浓缩,通过硅胶柱色谱(10∶1己烷/乙酸乙酯)纯化残余物,得到期望的作为白色固体的反式-α,β-不饱和的酯VII-c(32.0g,99%):
1H NMR(400MHz,CDCl3):δ8.45(d,J=16.0Hz,1H),8.29(dd,J=8.4,1.5Hz,1H),8.14(d,J=8.6Hz,1H),7.70(d,J=7.7Hz,1H),7.57(ddd,J=8.5,7.0,1.7Hz,1H),7.49(ddd,J=8.3,6.7,1.2Hz,1H),6.78(d,J=8.2Hz,1H),6.43(d,J=16.0Hz,1H),3.99(s,3H),3.83(s,3H).
化合物VII-d的制备
在室温下,使化合物VII-c(32.0g,132mmol)和10%Pd/C(5.0g)的EtOAc(400mL)混悬液处于氢化条件(1atm)16h。反应混合物通过硅藻土过滤并用MeOH洗涤。在真空下浓缩滤液,得到作为白色固体的VII-d(32g,99%):
1H NMR(400MHz,CDCl3):δ8.31(d,J=8.2Hz,1H),7.94(d,J=8.6Hz,1H),7.54(ddd,J=8.4,7.0,1.5Hz,1H),7.47(ddd,J=8.3,6.9,1.3Hz,1H),7.24(d,J=8.2Hz,1H),6.83(d,J=7.7Hz,1H),3.97(s,3H),3.68(s,3H),3.33(t,J=7.6Hz,2h),2.72(t,J=7.7Hz,2H).
化合物VII-e的制备
向甲酯VII-d(32.0g,131mmol)的THF/MeOH/H2O(200mL/200mL/75mL)的溶液加入NaOH(31.5g,786mmol)并将反应混合物在室温下搅拌1h。除去溶剂并用1N HCl水溶液将pH调整到1;过滤所沉淀的白色固体,用水洗涤并在真空下干燥,得到作为白色固体的酸VII-e(29.5g,98%):
1H NMR(400MHz,DMSO-d6):δ12.15(brs,1H),8.19(dd,J=8.6,1.1Hz,1H),7.99(d,J=8.4Hz,1H),7.57(ddd,J=8.2,7.9,1.3Hz,1H),7.50(ddd,J=8.4,7.1,1.5Hz,1H),7.28(d,J=7.9Hz,1H),6.88(d,J=8.1Hz,1H),3.94(s,3H),3.21(t,J=7.6Hz,2H),2.66(t,J=7.7Hz,2H).
化合物VII-g的制备
在-78℃下,向化合物VII-f(26.8g,151mmol)的无水THF(300mL)溶液逐滴加入正丁基锂(76.0mL,环己烷中的2M溶液)并且将反应混合物搅拌1h以得到化合物VII-f锂盐的溶液。在-78℃下,向化合物VII-e(29g,126mmol)的无水THF(300mL)的另一种溶液逐滴加入NMM(20.7mL,189mmol)和PivCl(18.6mL,151mmol)。将反应混合物在相同温度下搅拌1分钟并且于-78℃下缓慢添加所制备的化合物VII-f溶液。将反应混合物再搅拌10分钟,0℃下搅拌1h,在室温下搅拌30分钟,用饱和NH4Cl淬灭,浓缩以除去THF,并且在CH2Cl2(1000mL)与水(1000mL)之间分配。将水层分离并用CH2Cl2(2×1000mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过柱色谱(硅胶,CH2Cl2)纯化,得到作为白色固体的化合物VII-g(16g,33%):
1H NMR(400MHz,CDCl3):δ8.30(dd,J=8.6,1.3Hz,1H),8.02(d,J=8.2Hz,1H),7.55(ddd,J=8.5,6.9,1.6Hz,1H),7.47(ddd,J=8.2,6.9,1.2Hz,1H),7.34-7.22(m,5H),7.17(d,J=7.7Hz,1H),6.73(d,J=8.0Hz,1H),4.72-4.60(m,1H),4.14(d,J=2.4Hz,1H),4.13(s,1H),3.47-3.25(m,5H),3.96(s,3H),2.76(dd,J=13.2,9.6Hz,1H).
化合物VII-h的制备
在-78℃下,向化合物VII-g(16.0g,41.1mmol)的无水THF(500mL)的溶液中分批加入KHMDS(12.8g,61.7mmol)。将所得混合物搅拌30分钟之后,添加三异丙基苯磺酰叠氮化物(19.0g,61.7mmol)并将反应混合物搅拌5分钟。在相同温度下缓慢添加乙酸(24.7mL,411mmol),随后添加四甲基乙酸铵(10.9g,82.2mmol)。将反应混合物加热至24℃,搅拌16h,用饱和NaHCO3(300mL)淬灭,浓缩以除去THF,并用CH2Cl2(2×500mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩。残余物通过柱色谱(硅胶,90∶10己烷/EtOAc,随后是CH2Cl2)纯化,得到作为黄色固体的化合物VII-h(13.0g,74%):
1H NMR(400MHz,CDCl3):δ8.29(d,J=8.6Hz,1H),8.05(d,J=8.9Hz,1H),7.56(ddd,J=8.1,6.7,1.2Hz,1H),7.46(ddd,J=8.2,6.9,1.2Hz,1H),7.38(d,J=8.1Hz,1H),7.33-7.20(m,3H),7.18-7.12(m,2H),6.75(d,J=8.2Hz,1H),5.47(dd,J=8.4,7.0Hz,1H),4.44-4.35(m,1H),4.02(dd,J=9.2,2.7Hz,1H),3.93(s,3H),3.74(t,J=8.4Hz,1H),3.66(dd,J=1.41,7.0Hz,1H),3.46(dd,J=14.3,8.5Hz,1H),3.24(dd,J=13.4,3.4Hz,1H),2.75(dd,J=13.6,9.7Hz,1H).
化合物VII-i的制备
在0℃下,向化合物VII-h(26.0g,60.9mmol)的THF/H2O(100mL/35mL)溶液中加入H2O2(41.4mL,366mmol),随后分批添加LiOH(5.11g,122mmol)。将反应混合物搅拌10分钟,在室温下搅拌1h,用饱和Na2SO3(200mL)淬灭,减压下浓缩以除去THF,并用CH2Cl2(500mL)洗涤。水层用1N HCl水溶液酸化并用CH2Cl2(2×500mL)萃取。合并的有机萃取物经Na2SO4干燥,浓缩并用MTBE研磨,得到作为灰白色固体的化合物VII-i(13.5g,82%):
1H NMR(400MHz,DMSO-d6):δ8.22(dd,J=8.4,1.4Hz,1H),8.03(d,J=8.4Hz,1H),7.60(ddd,J=8.2,6.8,1.4Hz,1H),7.52(ddd,J=8.2,6.8,1.3Hz,1H),7.35(d,J=8.0Hz,1H),6.92(d,J=8.0Hz,1H),4.36(dd,J=9.5,5.0Hz,1H),3.96(s,3H),3.59(dd,J=14.9,5.1Hz,1H),3.25(dd,J=14.7,8.9Hz,1H).
化合物VII-j的制备
在室温下,使化合物VII-ji(13.5g,49.6mmol)和10%Pd/C(1.35g)的AcOH/H2O(300mL/100mL)混悬液处于氢化条件(1atm)3h。反应混合物通过硅藻土来过滤并用AcOH/H2O洗涤,随后用MeOH洗涤。在真空下浓缩滤液,得到作为黄色固体的乙酸盐VII-j(12.0g,80%):
ESI-MS m/z 246[C14H15NO3+H]+
化合物VII-k的制备
在室温下,向化合物VII-j(12.0g,39.3mmol)的乙酸(130mL)溶液逐滴加入氢溴酸(130mL)并且将反应混合物回流3h。将反应混合物冷却至室温并浓缩。将粗制棕色残余物VII-k(10.5g,86%)无需任何纯化直接用于下一个步骤:
1H NMR(400MHz,DMSO-d6):δ10.17(brs,1H),8.32(brs,3H),8.20(dd,J=8.3,1.5Hz,1H),8.00(d,J=8.5Hz,1H),7.57(ddd,J=8.2,6.7,1.2Hz,1H),7.49(ddd,J=8.2,6.8,1.1Hz,1H),7.21(d,J=7.7Hz,1H),6.82(d,J=7.7Hz,1H),4.11-3.98(m,1H),3.51-3.36(m,2H).
化合物VII-l的制备
在0℃下,将乙酰氯(16.8mL,236mmol)添加至无水甲醇(250mL)中,随后加入化合物VII-k(10.5g,33.7mmol)。将反应混合物回流4h并浓缩。残余物在CH2Cl2(500mL)与饱和NaHCO3(300mL)之间分配。将水层分离并用CH2Cl2(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩,得到作为白色固体的化合物VII-l(7.5g,91%):
1H NMR(400MHz,DMSO-d6):δ9.97(brs,1H),8.16(d,J=8.4Hz,1H),7.95(d,J=8.6Hz,1H),7.52(ddd,J=8.1,6.9,1.2Hz,1H),7.43(t,J=6.7Hz,1H),7.10(d,J=7.9Hz,1H),6.76(d,J=7.9Hz,1H),3.62(t,J=6.8Hz,1H),3.51(s,3H),3.37-3.26(m,2H),3.22(dd,J=14.3,6.7Hz,1H),3.08(t,J=14.3,7.7,Hz,1H).
化合物VII-m的制备
在0℃下,向化合物VII-l(7.5g,30.6mmol)的MeOH/H2O(300mL/100mL)溶液中加入NaHCO3(25.7g,306mmol)和Boc2O(10.0g,45.9mmol)。将所得的混合物加热至室温并搅拌1h。反应混合物在CH2Cl2(200mL)与水(200mL)之间分配。将水层分离并用CH2Cl2(2×400mL)萃取。合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩。使用20%乙酸乙酯/己烷随后使用CH2Cl2的快速柱色谱,得到作为白色固体的化合物VII-m(9.6g,91%):
1H NMR(300MHz,CDCl3):δ8.23(d,J=8.2Hz,1H),7.98(d,J=8.2Hz,1H),7.57-7.44(m,2H),7.07(d,J=8.0Hz,1H),6.68(d,J=7.6Hz,1H),6.55(brs,1H),5.14-4.85(brs,1H),4.77-4.51(m,1H),3.78-3.31(m,5H),1.40(s,6H),1.10(s,3H).
化合物VII-n的制备
在0℃下,向化合物VII-m(12.9g,37.5mmol)的吡啶(100mL)溶液中加入三氟甲磺酸酯(9.5mL,56.3mmol),并将反应混合物在室温下搅拌2h。浓缩之后,反应混合物在CH2Cl2(100mL)与水(50mL)之间分配。将水层分离并用CH2Cl2(2×50mL)萃取。合并的有机萃取物用盐水洗涤,经Na2SO4干燥,并浓缩,得到作为棕色油状物的化合物VII-n(22.0g,粗制):
1H NMR(400MHz,CDCl3):δ8.19-8.07(m,2H),7.69-7.64(m,2H),7.38(d,J=8.1Hz,1H),7.28(d,J=7.9Hz,1H),5.12-5.06(brs,1H),4.78-4.67(m,1H),3.68-3.46(m,5H),1.39(s,8H),1.25(s,1H).
化合物VII-p的制备
在室温下,向化合物VII-n(22.0g,粗制,37.51mmol)的无水CH3CN(250mL)溶液中加入TEA(20.5mL,150mmol)、己烷中的10%(t-Bu)3P(15.0mL,7.50mmol)、丁-3-炔基氨基甲酸苄酯(VII-o,11.3g,56.3mmol)和CuI(357mg,1.87mmol)。将所得的混合物用氩气脱气10分钟并一次性迅速地加入Pd(PPh3)4(4.33mg,3.75mmol)。用氩气脱气5分钟之后,将所得的混合物回流16h。在真空下浓缩反应混合物并且将残余物通过柱色谱(硅胶,60∶40乙酸乙酯/己烷)纯化,得到作为棕色油状物的化合物VII-p(14.0g,71%经过两个步骤):
1H NMR(400MHz,CDCl3):δ8.33(dd,J=7.5,2.2Hz,1H),8.07(dd,J=7.5,2.2Hz,1H),7.58-7.51(m,2H),7.52(d,J=7.5Hz,1H),7.35-7.29(m,5H),7.19(d,J=7.5Hz,1H),5.16-5.12(m,1H),5.13(s,2H),5.07-4.99(m,1H),4.74-4.65(m,1H),3.59(s,3H),3.91-3.42(m,2H),3.53(d,J=6.2Hz,2H),2.79(t,J=6.4Hz,2H),1.39(s,8H),1.25(s,1H).
化合物VII-q的制备
向甲酯VII-p(14.0g,26.5mmol)的THF(150mL)、甲醇(150mL)和水(75mL)混合物的溶液中加入固体NaOH(6.33g,159mmol)并且将反应混合物在室温下搅拌2h。当反应混合物的TLC显示反应完成时,将反应混合物的pH通过添加1N HCl(水溶液)恢复至9至10并除去有机溶剂。将水性部分的pH调整至5至6,并且所得的沉淀物用二氯甲烷来萃取。水性部分用CH2Cl2(2×50mL)萃取。合并有机层,经Na2SO4干燥并浓缩,得到作为棕色固体的化合物VII-q(13.0g,95%):
1H NMR(400MHz,DMSO-d6):δ8.32(d,J=7.4Hz,1H),8.13-8.05(m,1H),7.58-7.48(m,4H),7.38-7.29(m,5H),5.21-5.15(m,1H),5.12(s,2H),5.07-4.93(m,1H),4.70-4.54(m,1H),3.77-3.62(m,1H),3.57-3.35(m,2H),2.84-2.68(m,2H),1.37(s,9H).
化合物VII-r的制备
在冰浴中,将酸VII-q(4.00g,7.7mmol)的THF(100mL)溶液冷却至0℃。添加NMM(1.10mL,23.2mmol),随后添加PivCl(1.10mL,9.30mmol),并且将反应混合物在室温下搅拌2h。添加NH3(甲醇中的7.0N,11.0mL,77.5mmol)并将反应混合物在0℃下搅拌2h,恢复至室温并搅拌16h。除去有机溶剂。向残余物中加入水并用CH2Cl2(3×300mL)萃取。合并有机层,经Na2SO4干燥,过滤并浓缩。残余物通过柱色谱(氯仿中的3%甲醇)纯化,得到作为淡黄色固体的酰胺VII-r(3.50g,88%):
1H NMR(400MHz,DMSO-d6)8.29(d,J=7.1Hz,1H),8.21(d,J=7.0Hz,1H),7.63-7.52(m,4H),7.36-7.30(m,6H),6.91(d,J=7.6Hz,1H),5.05(s,2H),4.23-4.22(m,1H),3.61-3.60(m,1H),3.53-3.48(m,1H),3.16-3.06(m,1H),2.74-2.68(m,2H),1.24(s,9H).
化合物VII-s的制备
在室温下,使化合物VII-r(3.50g,6.8mmol)和10%Pd/C(700mg)的EtOH(100mL)和AcOH(20mL)的混悬液处于氢化条件(1atm)16h。反应混合物通过硅藻土过滤并用EtOH洗涤。在真空下浓缩滤液并用MTBE/己烷研磨,得到作为灰白色固体的乙酸盐VII-s(3.0g,99%):
1H NMR(400MHz,DMSO-d6)8.24-8.21(m,1H),8.10(d,J=7.0Hz,1H),7.56-7.54(m,2H),7.31-7.26(m,2H),4.44-4.42(m,1H),3.75-3.68(m,1H),3.64-3.59(m,1H),3.24-3.19(m,2H),3.13(t,J=6.2Hz 2H),2.92(t,J=6.2Hz,2H),1.84-1.71(m,4H),1.39(s,9H).
化合物VII-t的制备
在0℃下,向胺VII-s(3.0g,6.74)的MeOH(100mL)和水(50mL)溶液加入Na2CO3(7.14g,67.4mmol)并搅拌10分钟。在相同温度下添加氯甲酸苄酯(1.93mL,13.4mmol)并将反应混合物搅拌1h,恢复至室温,并且再搅拌1h。浓缩混合物,将残余物溶于CH2Cl2(200mL)中,该溶液用水(300mL)和盐水(300mL)洗涤。有机层经Na2SO4干燥,过滤并浓缩。残余物通过柱色谱纯化(氯仿中的3%甲醇)得到作为淡黄色固体的酰胺VII-t(3.10g,89%):ESI-MS m/z 520[C30H37N3O5+H]+。
化合物VII-u的制备
在室温下,将化合物VII-t(3.10g,5.80mmol)溶于二氧六环中的4N HCl(40mL)中并将该溶液搅拌1h。浓缩之后,获得为白色固体的胺盐VII-u(2.6g,99%),并且直接用于下一个步骤:
ESI-MS m/z 420[C25H29N3O3+H]+.
化合物2的制备
将1(10g)的CH2Cl2(100mL)溶液冷却至0℃。在10分钟之后,添加戴斯-马丁高碘烷(29g)并将反应混合物在室温下搅拌2h。添加1N NaOH(水溶液)并用CH2Cl2(3×300mL)萃取。合并有机层,经Na2SO4干燥,过滤并浓缩,得到作为淡黄色液体的醛2(8.0g,80%)。
化合物VII-v的制备
添加胺盐VII-u(3.70g,8.13mmol)、醛(2)(1.7g,9.75mmol)和乙酸(4.88mL)的溶液并在室温下搅拌10分钟。添加氰基硼氢化钠(768mg,12.2mmol)并将混合物搅拌2h。在30分钟内,加入额外的2(0.3当量)、AcOH(0.3当量)和NaCNBH3(0.3当量)。将反应混合物浓缩至干并且残余物用饱和NaHCO3(200mL)洗涤并用EtOAc(3×300mL)萃取。有机层经Na2SO4干燥、过滤并浓缩。该粗制产物(VII-v,8.0g)无需进一步纯化直接用于下一个步骤,并且产物组成通过LCMS数据来确认:ESI-MS m/z 577[C33H44N4O5+H]+。
化合物VII-w的制备
在0℃下,向胺VII-v(粗制产物8.0g)的MeOH(90mL)和水(30mL)溶液加入NaHCO3(6.82g,81.3mmol)并搅拌10分钟。添加氯甲酸苄酯(2.47mL)并将反应混合物在室温下搅拌1h,恢复至室温并再搅拌1h。浓缩混合物,将残余物溶解在CH2Cl2(200mL)中,并将该溶液用水(300mL)和盐水(300mL)洗涤。有机层经Na2SO4干燥、过滤并浓缩。该粗制产物VII-w(10.0g)无需进一步纯化直接用于下一个步骤,并且产物组成通过LCMS数据来确认:ESI-MS m/z711[C41H50N4O7+H]+。
化合物VII-x的制备
在室温下将化合物VII-w(粗制产物,10.0g)溶解在二氧六环中的4N HCl(25mL)中并且将溶液搅拌1h。浓缩之后,胺盐用NaHCO3水溶液中和。残余物通过柱色谱(氯仿中的6%甲醇)纯化,得到作为淡黄色固体的胺VII-x(2.50g,经过三个步骤为50%):
ESI-MS m/z 611[C36H42N4O5+H]+。
化合物VII-aa和化合物VII-z的制备
向胺VII-x(2.50g,4.09mmol)的甲醇(50mL)溶液中依次加入三醇(VII-y)(4.39g,16.4mmol)和乙酸(2.45mL)并在室温下搅拌10分钟。添加氰基硼氢化钠(1.54mg,24.5mmol)并将混合物在室温下搅拌24h。加入额外的VII-y(2.0当量)、AcOH(4.0当量)和NaCNBH3(3.0当量)并且将该混合物搅拌48h。LC/MS显示90%胺的消耗。再次添加VII-y(2.0当量)、AcOH(4.0当量)和NaCNBH3(3.0当量)并且将该混合物搅拌24h。向反应混合物中加入己醛(1.46mL,12.2mmol)和NaCNBH3(1.26g,20.0mmol),搅拌2h并浓缩至干。残余物用饱和NaHCO3(200mL)洗涤并用EtOAc(3×300mL)萃取。有机层经Na2SO4干燥、过滤并浓缩。化合物VII-aa和化合物VII-z的纯化使用CMA系统的标准色谱来进行;使用C18Gold柱的反相色谱分别用于得到纯的VII-z(810mg,21%)和VII-aa(1.10g,25%):ESI-MS m/z947[C55H70N4O10+H]+对应于VII-z和ESI-MS m/z 1115[C62H74N4O15+H]+对应于VII-aa。
化合物VII-bb的制备
使VII-aa(1.10g,0.98)和10%Pd/C(200mg)的EtOH(80mL)和AcOH(20mL)混合物混悬液进行脱气并在室温下处于氢化条件(1atm)16h。反应混合物通过硅藻土垫来过滤并用MeOH洗涤。在真空下浓缩滤液,得到作为白色固体的胺盐VII-bb(925mg,92%):
ESI-MS m/z 847[C46H62N4O11+H]+。
化合物VII-dd的制备
在室温下,向胺盐VII-bb(925mg,0.95mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯(VII-cc,561mg,1.44mmol)的EtOH(10mL)溶液中逐滴加入DIPEA(1.35mL,7.60mmol)。在密封管中,于70℃下将反应混合物加热2h,冷却至室温并在真空中浓缩。残余物通过柱色谱(硅胶,80∶18∶2CHCl3/CH3OH/NH4OH)纯化,得到作为黄色固体的胍VII-dd(500mg,50%):
1H NMR(400MHz,CD3OD):δ8.21-8.16(m,1H),8.12-8.07(m,1H),7.56-7.49(m,2H),7.45-7.39(m,4H),7.32-7.22(m,8H),5.45(s,2H),4.21(dd,J=10.5,5.3Hz,2H),3.98-3.89(m,4H),3.83(dd,J=4.9,2.5Hz,2H),3.68(dd,J=9.2,2.5Hz,2H),3.57(t,J=10.5Hz,2H),3.45-3.37(m,2H),3.27(t,J=7.7Hz,2H),3.24-3.17(m,1H),3.15-3.08(m,2H),2.64-2.44(m,6H),2.43-2.25(m,2H),1.88-1.79(m,2H),1.79-1.68(m,2H),1.54-1.41(m,2H).
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(化合物VII-ee)的制备:
将4N HCl水溶液(20mL)添加至VII-dd(500mg,0.48mmol)并且在室温下将反应混合物搅拌2h。除去溶剂并使用C18Gold柱通过反相色谱纯化残余物,得到作为黄色吸湿性固体的盐酸盐VII-ee(285mg,60%):
1H NMR(400MHz,DMSO-d6):δ10.53(brs,1H),10.29(brs,1H),9.46(brs,1H),9.32(brs,1H),9.05-8.78(m,3H),8.41-8.33(m,1H),8.17-8.09(m,1H),7.74(s,1H),7.58(dd,J=6.4,3.2Hz,2H),7.51(s,1H),7.48-7.35(m,1H),7.32(d,J=7.8Hz,1H),7.28(d,J=7.3Hz,1H),4.16-4.05(m,2H),4.04-3.96(m,1H),3.84(dd,J=12.8,3.3Hz,1H),3.72(d,J=5.8Hz,2H),3.62(d,J=2.8Hz,1H),3.58(d,J=2.8Hz,1H),3.54-3.45(m,4H),3.44-3.30(m,9H),3.28-3.18(m,2H),3.15-3.01(m,2H),2.99-2.85(m,2H),2.28-2.13(m,2H),1.81-1.63(m,4H).
1H NMR(400MHz,CD3OD):δ8.25(dd,J=8.5,1.7Hz,1H),8.16(dd,J=8.1,1.5Hz,1H),7.64-7.54(m,2H),7.37(ABq,J=7.4Hz,2H),4.25-4.17(m,3H),3.88-3.84(m,2H),3.83(dd,J=13.8,4.8Hz,1H),3.79(d,J=2.8Hz,1H),3.76(d,J=2.8Hz,1H),3.73-3.63(m,5H),3.61-3.50(m,3H),3.49-3.42(m,4H),3.41-3.35(m,3H),3.23-3.06(m,4H),2.36-2.23(m,2H),1.93-1.77(m,4H).
方案VIII.3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(VIII-d)的制备
化合物VIII-b的制备
在室温下使VII-z(800mg,0.86)和10%Pd/C(160mg)在EtOH(80mL)和AcOH(20mL)混合物中的混悬液脱气并置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用MeOH洗涤。将滤液在真空下浓缩,得到作为白色固体的胺盐VIII-b(670mg,91%):
ESI-MS m/z 679[C39H58N4O6+H]+.
化合物VIII-d的制备
在室温下向胺盐VIII-b(670mg,0.78mmol)和3,5二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯(VIII-a,485mg,1.24mmol)的EtOH(10mL)溶液中添加DIPEA(1.10mL,6.24mmol)。将反应混合物在密封管中于70℃下加热2h,冷却至室温并在真空下浓缩。通过柱色谱(硅胶,80∶18∶2CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的胍VIII-c(360mg,52%):
1H NMR(400MHz,CD3OD):δ8.24-8.18(m,1H),8.14-8.07(m,1H),7.56-7.48(m,2H),7.47-7.40(m,2H),7.32-7.22(m,5H),5.50(s,1H),4.23(dd,J=10.8,5.8Hz,1H),4.00-3.91(m,1H),3.86(dd,J=5.4,1.9Hz,1H),3.73(dd,J=9.5,2.5Hz,1H),3.59(t,J=10.8Hz,2H),3.46-3.37(m,2H),3.12(t,J=6.8Hz,2H),3.24-3.17(m,1H),2.67(dd,J=13.8,4.5Hz,1H),2.55-2.26(m,8H),1.90-1.70(m,4H),1.51-1.41(m,2H),1.36-1.08(m,9H),0.86(t,J=7.2Hz,3H).
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(化合物VIII-d)的制备:
向VIII-dc(360mg,0.40mmol)中添加4N HCl水溶液(20mL),并在室温下将反应混合物搅拌2h。除去溶剂并使用C18Gold柱通过反相色谱纯化残余物,得到作为黄色吸湿性固体的盐酸盐VIII-d(70mg,36%):
1H NMR(400MHz,DMSO-d6):δ10.52(s,1H),10.49-10.28(m,1H),9.72-9.55(m,1H),9.47-9.34(m,1H),9.29(brs,1H),9.01-8.74(m,2H),8.43-8.35(m,1H),8.20-8.11(m,1H),7.73(s,1H),7.62-7.55(m,2H),7.52(s,1H),7.47-7.37(m,2H),7.32(d,J=7.1Hz,1H),7.27(d,J=7.6Hz,1H),3.73-3.67(m,2H),3.60(dd,J=10.6,3.0Hz,1H),3.55-3.45(m,2H),3.44-3.22(m,3H),3.31-3.22(m,3H),3.20-3.00(m,5H),2.98-2.86(m,2H),2.24-2.08(m,2H),1.79-1.61(m,6H),1.36-1.22(m,6H),0.88(t,J=6.4Hz,3H).
1H NMR(400MHz,CD3OD):δ8.26(d,J=8.4Hz,1H),8.16(dd,J=8.1,1.3Hz,1H),7.63-7.54(m,2H),7.37(q,J=7.2Hz,2H),4.22-4.13(m,2H),3.89-3.81(m,2H),3.77(dd,J=10.6,3.1Hz,1H),3.73-3.64(m,3H),3.55-3.49(m,1H),3.49-3.46(m,1H),3.43-3.34(m,6H),3.27-3.07(m 5H).2.32-2.17(m,2H),1.93-1.71(m,6H),1.46-1.33(m,6H),0.93(t,J=6.4Hz,3H).
方案IX
3,5-二氨基-N-(N-(4-(6-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺
化合物IX-b的制备
在室温下使IX-a(5.70g,11.0mmol)和10%林德拉催化剂(1.0g)在EtOH(100mL)和THF(20mL)中的混悬液置于氢化条件(1atm)下36h。将反应混合物用硅藻土的塞子过滤且塞子用MeOH洗涤。将滤液在真空下浓缩,并通过柱色谱(硅胶,95∶5CHCl3/CH3OH)纯化残余物,得到作为黄色固体的化合物IX-b(5.20g,92%):
ESI-MS m/z 518[C30H35N3O5+H]+。
化合物IX-c的制备
在室温下将化合物IX-b(5.20g,10.0mmol)溶解于二氧六环中的4N HCl(40mL)中并将溶液搅拌2h。浓缩后,得到作为白色固体的胺盐IX-c(4.50g,99%):ESI-MS m/z 418[C25H27N3O3+H]+419.
3-氧代丙基氨基甲酸叔丁酯2的制备
将1(10g)的CH2Cl2(100mL)溶液冷却至-0℃,10分钟后,添加戴斯马丁高碘烷(29g),并在室温下将反应混合物搅拌2h。添加1N NaOH(水溶液)并用CH2Cl2(3×300mL)萃取。将有机层合并,经Na2SO4干燥、过滤并浓缩,得到作为浅黄色液体的醛2(9.0g,91%)并直接用于接下来的步骤。
化合物IX-d的制备
向胺盐IX-c(4.50g,10.0mmol)的甲醇(100mL)溶液中添加醛(2)(2.0g,12.0mmol)的乙酸(6.0mL)溶液,并在室温下搅拌10分钟,然后添加氰基硼氢化钠(942mg,15.0mmol)并在室温下搅拌2h。在2h的时间内,添加额外的2(0.3当量)、AcOH(0.5当量)、NaCNBH3(0.5当量),并将这样的添加反复进行3次,直到LC-MS显示>90%的胺消耗。将反应混合物浓缩至干燥,用饱和NaHCO3(200mL)洗涤残余物,并用EtOAc(3×300mL)萃取。使有机层经Na2SO4干燥、过滤、浓缩。通过LC-MS分析确认粗制品IX-d(8.0g)且所述粗制品无需进一步纯化直接用于接下来的步骤:ESI-MS m/z 575[C33H42N4O5+H]+.
化合物IX-e的制备
在0℃向在MeOH(150mL)和水(50mL)中的胺1X-d(粗制品,8.0g)的溶液中添加NaHCO3(8.40g,100mmol)并搅拌10分钟,然后在相同的温度下逐滴添加氯甲酸苄酯(3.0mL,20.0mmol),并在相同的温度下将反应混合物搅拌2h,然后升至室温,再搅拌1h。将混合物浓缩,使残余物溶解于CH2Cl2(200mL)中,并用水(300mL)和盐水(300mL)洗涤溶液。将有机层经Na2SO4干燥、过滤并浓缩。通过LC-MS分析确认这种粗制品IX-e(18.0g)且所述粗制品无需进一步纯化直接用于接下来的步骤:ESI-MS m/z 709[C41H48N4O7+H]+。
化合物IX-f的制备
在室温下将化合物IX-e(粗制品,18.0g)溶解于二氧六环中的4N HCl(50mL)中并将溶液搅拌2h。浓缩后,用NaHCO3水溶液中和胺盐。通过柱色谱(氯仿中的6%甲醇)纯化残余物,得到作为浅黄色固体的胺IX-f(2.50g,经三步的产率41%):ESI-MS m/z609[C36H40N4O5+H]+。
化合物IX-h和IX-i的制备
在室温下向胺IX-f(2.50g,4.10mmol)的甲醇(100mL)溶液中依次添加三醇(IX-g)(3.30g,12.3mmol)、乙酸(2.46mL)并搅拌10分钟,然后添加氰基硼氢化钠(1.30g,20.5mmol),并在室温下搅拌16h。在16h的时间内,添加额外的IX-g(2.0当量)、AcOH(5.0当量)、NaCNBH3(3.0当量),并将这样的添加再重复一次并搅拌16h。向此反应混合物中添加己醛(1.47mL,12.3mmol)、AcOH(0.7mL)、NaCNBH3(774mg)并搅拌1h,将反应混合物浓缩至干燥,用饱和NaHCO3(200mL)洗涤残余物,用EtOAc(3×300mL)萃取。使有机层经Na2SO4干燥、过滤、浓缩。使用CMA系统通过正常的色谱未能纯化化合物IX-h和IX-i,然后使用C-18Gold反相柱分别得到纯的IX-h(1.30g,34%)和IX-i(1.43g,28%):对于IX-h为ESI-MS m/z 945[C55H68N4O10+H]+.且对于IX-i为ESI-MS m/z 1113[C62H72N4O15+H]+。
化合物IX-j的制备
在室温下使IX-h(1.3g,1.37)和10%Pd/C(400mg)在EtOH(100mL)和AcOH(30mL)混合物中的混悬液脱气,然后置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤且塞子用MeOH洗涤。将滤液在真空下浓缩,得到作为白色固体的胺盐IX-j(1.15g,98%):ESI-MS m/z 679[C39H58N4O6+H]+679。
化合物IX-l的制备
在室温下向胺盐IX-j(1.15g,1.34mmol)和甲基3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯(IX-k,834mg,2.14mmol)的EtOH(20mL)溶液中添加DIPEA(1.90mL,10.72mmol)。将反应混合物在密封管中于70℃下加热2h,然后冷却至室温并在真空中浓缩。通过柱色谱(硅胶,80∶18∶2CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的胍IX-l(800mg,67%):
1H NMR(400MHz,CD3OD):δ7.72(dd,J=8.3,2.7Hz,2H),7.62(d,J=86Hz,2H),7.44(d,J=7.4,3.9Hz,2H),7.38-7.32(m,2H),7.32-7.27(m,3H),5.51(s,1H),4.25(dd,J=10.8,5.8Hz,1H),4.04-3.93(m,2H),3.89(dd,J=5.3,1.7Hz,1H),3.74(dd,J=9.2,2.0Hz,1H),3.61(t,J=11.0Hz,1H),3.45(t,J=7.1Hz,1H),3.17-3.05(m,1H),2.95(dd,J=13.8,7.4Hz,1H),2.83(t,J=7.1Hz,2H),2.80-2.69(m,3H),2.67-2.56(m,2H),2.54-2.44(m,3H),1.87-1.78(m,2H),1.76-1.67(m,2H),1.64-1.54(m,2H),1.36-1.17(m,6H),1.16-1.01(m,4H),0.85(t,J=7.3Hz,3H).
3,5-二氨基-N-(N-(4-(6-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(化合物IX-m)的制备:
将4N HCl水溶液(25mL)添加到IX-l(800mg,0.89mmol)中并在室温下将反应混合物搅拌2h。除去溶剂,使用C-18Gold柱通过反相柱纯化残余物,得到作为黄色吸湿性固体的盐酸盐IX-m(500mg,62%):
1H NMR(400MHz,DMSO-d6)10.51(brs,1H),9.29(brs,1H),8.94(brs,1H),8.82(brs,1H),7.77(dd,J=8.7,3.1Hz,2H),7.68(d,J=10.7Hz,2H),7.59(brs,1H),7.47-7.34(m,4H),7.17(brs,1H),5.97-5.09(m 3H),4.64(brs,1H),4.44(brs,1H),3.96(brs,1H),3.69(d,J=11.1Hz,1H),3.56-3.49(m,1H),3.48-3.40(m,2H),3.17-2.90(m,6H),2.84-2.66(m,1H),2.78(t,J=7.5Hz,2H),1.75-1.67(m,2H),1.65-1.55(m,2H),1.30-1.07(m,6H),0.85(t,J=7.2Hz,3H)
1H NMR(400MHz,CD3OD)7.77(d,J=8.5Hz,2H),770(s,1H),7.65(s,1H),740(dt,J=8.6,1.7Hz,2H),4.15-4.08(m,1H),3.83-3.77(m,2H),3.76-3.64(m,4H),3.37(t,J=7.1Hz,2H),3.24(d,J=10.6Hz,2H),3.20-3.08(m,4H),2.85(t,J=7.4Hz,2H),2.97-2.76(m,5H),1.95-1.80(m,4H),1.79-1.70(m,2H),1.55(brs,1H),1.35-1.12(m,6H),1.45-1.36(m,1H),0.90(t,J=7.1Hz,3H).
方案X
3,5-二氨基-N-(N-(4-(6-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺
化合物X-b的制备
在室温下使X-a(1.43g,1.28)和10%Pd/C(400mg)在EtOH(100mL)和AcOH(40mL)混合物中的混悬液进行脱气,然后置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤且塞子用MeOH洗涤。将滤液在真空中浓缩,得到作为白色固体的胺盐X-b(1.30g,99%):ESI-MS m/z 847[C46H62N4O11+H]+。
化合物X-c的制备
在室温下向胺盐X-b(1.30g,1.26mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯(X-e,788mg,2.02mmol)的EtOH(20mL)溶液中添加DIPEA(1.79mL,10.0mmol)。将反应混合物在密封管中于70℃下加热2h,然后冷却至室温并在真空中浓缩。通过柱色谱(硅胶,80∶18∶2CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的胍X-c(900mg,68%):
1H NMR(400MHz,CD3OD):δ7.70(d,J=8.1Hz,2H),7.59(s,2H),7.46-7.39(m,4H),7.34-7.24(m,8H),5.45(s,2H),4.21(dd,J=10.7,5.5Hz,2H),3.99-3.88(m,6H),3.82(dd,J=5.3,2.4Hz,2H),3.67(dd,J=9.8,2.9Hz,2H),3.57(t,J=10.6,2H),3.23(t,J=6.7Hz,2H),3.06(dd,J=13.4,6.6Hz,1H),2.93(dd,J=13.6,7.5Hz,1H),2.79(t,J=7.6Hz,2H),2.63-2.45(m,5H),2.44-2.36(m,2H),1.84-1.73(m,2H),1.70-1.60(m,2H),1.57-1.44(m,2H).
3,5-二氨基-N-(N-(4-(6-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(化合物X-d)的制备:
将4N HCl水溶液(30mL)添加到X-c(900mg,0.85mmol)中并在室温下将反应混合物搅拌2h。除去溶剂,使用C-18Gold柱通过反相柱纯化残余物,得到作为黄色吸湿性固体的盐酸盐X-d(590mg,70%):
1H NMR(400MHz.DMSO-d6)10.51(brs,1H),9.81(brs,1H),9.29(brs,1H),8.94(brs,1H),8.83(brs,1H),7.94(brs,1H),7.80(brs,1H),7.78(brs,1H),7.71(brs,1H),7.68(brs,1h),7.59(brs,1H),7.46-7.35(m,4H).5.47(brs,2H),4.84(brs,1H),4.69-4.54(m,3H),4.43(brs,1H),4.18-3.98(m,3H),3.74-3.67(m,2H),3.63-3.61(m,1H),3.60-3.56(m,1H),3.55-3.38(m,6H),3.27-3.09(m,3H),3.02-2.86(m,2H),2.79(t,J=7.1Hz,2H),2.25-2.09(m,2H),1.79-1.67(m,2H),1.65-1.54(m,2H).
1H NMR(400MHz,CD3OD)7.79(brs,1H),7.77(brs,1H),7.74(brs,1H),7.66(brs,1H),7.40(dt,J=6.8,1.6Hz,2H),4.24-4.12(m,3H),3.86-3.82(m,2H),3.80(d,J=3.2Hz,1H),3.77(d,J=3.4Hz,1H),3.74-3.63(m,6H0,3.52-3.33(m,6H),3.36(t,J=6.9Hz,2H),3.28-3.21(m,2H),3.15-3.07(m,2H),2.86(t,J=7.1Hz,2H),2.28-2.04(m,2H),1.90-1.80(m,2H),1.79-1.70(m,2H).
1,4-四氢萘基酪氨酸衍生物的制备
方案XI
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺
化合物XI-c的制备
在0℃将TEA(340mL)缓慢添加到甲酸(150mL)中。添加TEA之后,添加化合物XI-a(76.4g,401.6mmol)和化合物XI-b(59.2g,415.0mL)。将反应混合物加热至回流12h,冷却至室温并倒入冰水(600mL)中。通过添加NaOH(70g)的水(1.4L)溶液将溶液的pH值调整至11。用EtOAc萃取(600mL 3次)所得的溶液并酸化至pH 2-3。在50℃过滤白色沉淀物并在真空下干燥,得到作为灰白色固体的化合物XI-c(74.6g,79%)。
1H NMR(400MHz,MeOD-d4):δ6.92(d,J=8.4Hz,1H),6.67(dd,J=8.4Hz,8.4Hz,1H),3.75(s,3H),2.81(t,J=7.6Hz,2H),2.67(t,J=6.0Hz,2H),2.62(t,J=6.0Hz,2H),2.49(t,J=7.6Hz,2H),1.78-1.73(m,4H).
化合物XI-e的制备
在-10℃向化合物XI-c(36g,153.8mmol)的无水THF(400mL)溶液中逐滴添加TEA(56mL,400.0mmol)和新戊酰氯(22.7mL,184.6mmol)。在-10℃将混合物搅拌40分钟,随后添加化合物XI-d(32.7g,184.6mmol)和LiCl(8.5g,184.6mmol)的THF(200mL)溶液。将反应混合物升温至室温,搅拌12h,用饱和NaHCO3洗涤,浓缩以除去THF,使混合物在EtOAc(1000mL)与水(1000mL)之间分配。分离出水层并用EtOAc(2×800mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩。将残余物从EtOAc/己烷中重结晶(3∶1,v/v),得到作为白色固体的化合物XI-e(40.7g,78%)。
1H NMR(400MHz,CDCl3):δ7.32-7.25(m,3H),7.20(d,J=8.0Hz,1H),7.02(d,J=8.0Hz,1H),6.63(d,J=8.0Hz,1H),4.68-4.62(m,1H),4.18-4.16(m,2H),3.79(s,3H),3.31(dd,J=13.6,3.2Hz,1H),3.23-3.16(m,2H),2.95-2.91(m,2H),2.78-2.71(m,3H),2.66(t,J=7.5Hz,2H),1.80-1.75(m,4H).
化合物XI-f的制备
在-78℃向化合物XI-e(25.0g,63.5mmol)在无水THF(500mL)中的溶液中分批添加KHMDS(18.0g,95.3mmol)。在将所得混合物搅拌30分钟后,添加三异丙基苯磺酰叠氮化物(25.0g,82.6mmol)并将反应混合物搅拌2-3分钟。在相同温度下添加乙酸(19.1g,317.5mmol),随后添加乙酸钾(31.0g,317.5mmol)。将反应混合物升温至27℃,搅拌16h,并用盐水(500mL)淬灭。分离出水层并用EtOAc(3×500mL)萃取。将合并的有机萃取物用饱和NaHCO3和盐水洗涤,经Na2SO4干燥并浓缩。通过柱色谱(硅胶,90∶10己烷/EtOAc)纯化残余物,得到作为无色油状物的合物XI-f(15.0g,60%)。
1H NMR(400MHz,CDCl3):δ7.34-7.28(m,3H),7.25-7.20(m,2H),7.01(d,J=8.4Hz,1H),6.61(d,J=8.4Hz,1H),5.36(t,J=7.6Hz,1H),4.55-4.45(m,1H),4.10(dd,J=9.2,2.4Hz,1H),3.94(t,J=8.4Hz,1H),3.78(s,3H),3.30(dd,J=11.2,3.2Hz,1H),3.12(dd,J=8.0,2.4Hz,2H),2.81-2.72(m,3H),2.63(t,J=6.4Hz,2H),1.80-1.73(m,4H).
化合物XI-g的制备
在0℃向化合物XI-f(22.0g,50.6mmol)的THF/H2O(450mL/150mL)溶液中添加H2O2(25mL,253mmol),随后分批添加LiOH(4.7g,111mmol)。在相同的温度下将反应混合物搅拌3h,用饱和Na2SO3(300mL)淬灭,在减压下浓缩以除去THF,并用CH2Cl2(200mL)洗涤。用2N HCl水溶液使水层酸化并用CH2Cl2(2×250mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩,得到作为灰白色固体的化合物XI-g(11.0g,79%)。粗制品无需纯化直接用于接下来的步骤。
化合物XI-h的制备
在室温下使化合物XI-g(10.0g,36.3mmol)和10%Pd/C(3.50g)在AcOH/H2O(200mL/50mL)中的混悬液置于氢化条件(1atm)下12h。将反应混合物通过硅藻土过滤并用MeOH洗涤。将滤液在真空下浓缩,得到作为黄色固体的乙酸盐XI-h(8.0g,88%)。粗制品无需纯化直接用于接下来的步骤。
化合物XI-i的制备
在室温下向化合物XI-h(13.0g,52.3mmol)的乙酸溶液(150mL)中逐滴添加40%氢溴酸(150mL)并将反应混合物回流4h。将反应混合物冷却至室温并浓缩。用H2O(15mL)稀释残余物,用氨水略微碱化并过夜结晶,得到作为棕色固体的化合物XI-i(15.0g,90%)。通过LC/MS表征产物且产物无需纯化即用于接下来的步骤。
ESI-MS m/z 236[C13H17NO3+H]+。
化合物XI-j的制备
在0℃将乙酰氯(26.0g,332mmol)添加到无水甲醇(210mL)中,随后添加化合物XI-i(15.0g,47.4mmol)。将反应混合物回流4h并浓缩。使残余物在CH2Cl2(300mL)和饱和NaHCO3(300mL)之间分配。分离出水层并用CH2Cl2(2×300mL)萃取。合并的有机萃取物经Na2SO4干燥并浓缩,得到作为无色油状物的化合物XI-j(15.0g,粗制品)。通过LC/MS表征粗制品且无需纯化即用于接下来的步骤。
ESI-MS m/z 250[C14H19NO3+H]+。
化合物XI-k的制备
在0℃向化合物XI-j(15.0g,47.0mmol)的MeOH/H2O(160mL/160mL)溶液中添加NaHCO3(17.0g,200.0mmol)和Boc2O(12.8g,60.0mmol)。将所得混合物升温至室温并搅拌3h。使反应混合物在CH2Cl2(200mL)与水(200mL)之间分配。分离出水层并用CH2Cl2(2×200mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为无色油状物的化合物XI-k(9.0g,从化合物XI-h经三步产率50%)。
1H NMR(400MHz,CDCl3):δ6.77(d,J=8.4Hz,1H),6.55(d,J=8.4Hz,1H),4.95(d,J=8.0Hz,1H),4.70(s,1H),4.50(t,J=6.5Hz,1H),3.69(s,3H),3.42(dd,J=14.0,6.0Hz,1H),2.89-2.84(m,1H),2.68-2.63(m,4H),1.79(t,J=3.2Hz,4H),1.40(s,9H).
化合物XI-l的制备
在0℃向化合物XI-k(9.40g,26.9mmol)的吡啶溶液(100mL)中添加三氟甲磺酸(11.4g,40.4mmol),并在室温下将反应混合物搅拌2h。浓缩后,使反应混合物在CH2Cl2(300mL)与水(300mL)之间分配。分离出水层并用CH2Cl2(2×300mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为棕色油状物的化合物XI-l(9.10g,71%)。1H NMR和LC-MS数据与产物相符。
1H NMR(400MHz,CDCl3):δ7.29-7.26(m,2H),5.04(d,J=7.8Hz,1H),4.56(d,J=7.2Hz,1H),3.68(s,3H),3.12-3.04(m,1H),2.95-2.90(m,1H),2.80-2.73(m,4H),1.83-1.79(m,4H),1.38(s,9H).
化合物XI-n的制备
在室温下向化合物XI-l(9.10g,18.9mmol)在无水CH3CN(100mL)中的溶液中添加TEA(7.6g,75.6mmol)、在己烷中的(t-Bu)3P(0.76g,3.78mmol)、丁-3-炔基氨基甲酸苄酯(XI-m,5.74g,28.3mmol)和CuI(180mg,0.94mmol)。将所得混合物用氩气脱气3分钟并一次性快速添加Pd(PPH3)4(2.18g,1.89mmol)。用氩气脱气5分钟后,将所得混合物回流4h。将反应混合物在真空下浓缩并通过柱纯化残余物,得到作为棕色油状物的化合物XI-n(7.50g,经两步的产率74%)。1H NMR和LC-MS的数据与产物相符。
1H NMR(300MHz,CDCl3):δ7.36-7.25(m,5H),7.15(d,J=7.8Hz,1H),6.83(d,J=7.8Hz,1H),5.12(br s,3H),4.97(d,J=7.6Hz,1H),4.52(d,J=6.8Hz,1H),3.67(s,3H),3.46-3.40(m,2H),3.11-3.04(m,1H),2.95-2.83(m,3H),2.68-2.64(m,4H),1.77-1.75(m,4H),1.39(s,9H).
化合物XI-o的制备
向甲酯XI-n(7.50g,14.04mmol)的THF/MeOH/H2O溶液(50mL/50mL/25mL)中添加NaOH(1.12g,28.08mmol)并在室温下将反应混合物搅拌1h。用1N HCl水溶液将pH值调节至9并除去该有机溶剂。将残余物的pH值调节至5且使混悬液在CH2Cl2(200mL)与水(200mL)之间分配。分离出水层并用CH2Cl2(2×200mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩,得到作为棕色固体的化合物XI-o(6.50g,90%)。
1H NMR(400MHz,DMSO-d6):δ7.44(t,J=5.8Hz,1H),7.33-7.28(m,5H),7.15(d,J=8.4Hz,1H),7.09(d,J=7.8Hz,1H),6.99(d,J=7.8Hz,1H),5.02(br s,2H),4.07-4.01(m,1H),3.61-3.58(m,1H),3.24-3.15(m,2H),3.02-2.97(m,1H),2.79-2.75(m,3H),2.66-2.64(m,2H),2.58(t,J=7.0,Hz,2H),1.73-1.69(m,4H),1.31(s,9H).
化合物XI-p的制备
在0℃向酸XI-o(6.50g,12.5mmol)的THF(200mL)溶液中添加NMM(1.89g,18.75mmol)和i-BCF(2.04g,15.0mmol)。在相同温度下将反应混合物搅拌1h,并逐滴添加NH3(在甲醇中7.0N,29.4mL,206mmol)。在0℃将反应混合物继续搅拌2h,升温至室温并搅拌1h。浓缩后,使残余物在CH2Cl2(100mL)与水(100mL)之间分配。分离出水层并用CH2Cl2(2×100mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。将残余物用MTBE洗涤,得到作为黄色固体的酰胺XI-p(5.90g,70%)。
1H NMR(400MHz,DMSO-d6):δ7.44(t,J=5.8Hz,1H),7.33-7.28(m,5H),7.15(d,J=8.4Hz,1H),7.09(d,J=7.8Hz,1H),6.99(d,J=7.8Hz,1H),5.02(s,2H),4.12-4.05(m,1H),3.62-3.53(m,1H),3.25-3.13(m,3H),2.94-2.88(m,1H),2.74-2.67(m,4H),2.60(t,J=7.0,Hz,2H),1.74-1.69(m,4H),1.31(s,9H).
化合物XI-q的制备
在室温下使化合物XI-p(5.90g,11.3mmol)和10%Pd/C(59mg)在EtOH(100mL)/AcOH(20mL)的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用EtOH洗涤。将滤液在真空下浓缩,并用MTBE/己烷洗涤,得到作为无色液体的乙酸盐XI-q(6.50g,粗制品)。ESI-MS m/z 390[C22H35N3O3+H]+.
化合物XI-r的制备
在0℃向化合物XI-q(6.50g,粗制品)在MeOH(300mL)/水(100mL)中的搅拌溶液中添加Na2CO3和CbzCl(4.20g,25.06mmol)并在相同温度下搅拌1h。在室温下将反应混合物搅拌1h,除去溶剂,在CH2Cl2(500mL)与水(100mL)之间分配。分离出水层并用CH2Cl2(2×100mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为黄色固体的化合物XI-r(3.90g,经两步的产率66%)。
1H NMR(300MHz,CDCl3):δ7.37-7.32(m,5H),6.96-6.89(m,2H),5.65(br s,1H),5.29(br s,1H),5.08(s,2H),4.71-4.69(m,1H),4.31-4.28(m,1H),3.21(t,J=6.2Hz,2H),3.11-2.95(m,2H),2.74(br s,2H),2.67(br s,2H),2.54(br s,2H),1.73-1.69(m,4H),1.40(s,9H).
化合物XI-s的制备
向化合物XI-r(3.90g,7.45mmol)的二氧六环溶液中添加二氧六环中的4N HCl溶液(30mL),并在室温下将反应混合物搅拌4h。在真空下除去溶剂,并将残余物用MTBE洗涤,得到作为黄色油状物的化合物XI-s(3.0g,95%)。
1H NMR(300MHz,CD3OD):δ7.33-7.25(m,5H),7.00-6.97(m,2H),5.05(s,2H),4.00-3.92(m,1H),3.20-2.98(m,4H),2.77-2.65(m,4H),2.57(br s,2H),1.81-1.77(m,4H),1.55-1.54(m,4H).
化合物XI-t的制备
向化合物XI-s(3.0g,7.09mmol)和醛2(1.47g,8.51mmol)在MeOH(100mL)中的溶液中添加乙酸(5.0mL),并在室温下将反应混合物搅拌10分钟。添加氰基硼氢化钠(670mg,10.63mmol)并将溶液在室温下继续搅拌1h。添加额外的化合物2(0.3当量)、AcOH(0.5当量)、和NaCNBH3(0.5当量)并搅拌1h。浓缩后,使残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。分离出水层并用EtOAc(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。残余物XI-t(3.50g,粗制品)无需进一步纯化即直接用于接下来的步骤。
化合物XI-u的制备
在0℃向化合物XI-t(3.50g,在MeOH/H2O(100mL/50mL)中的粗制品)的溶液中添加饱和Na2CO3,并将溶液搅拌10分钟。逐滴添加氯甲酸苄酯(1.53g,9.05mmol),并在0℃下将反应混合物搅拌1h,升温至室温并搅拌1h。浓缩后,残余物溶解于CH2Cl2(200mL)中,然后用水(300mL)和盐水(300mL)洗涤。将有机层经Na2SO4干燥并浓缩,通过柱纯化,得到作为黄色油状物的XI-u(3.20g,经两步的产率65%)。
1H NMR(300MHz,CD3OD):δ7.32-7.26(m,13H),6.91-6.73(m,3H),5.15-4.95(m,4H),4.48-4.31(m,1H),3.63-3.51(m,1H),3.17-3.03(m,9H),2.87-2.67(m,4H),2.57(br s,7H),1.76-1.63(m,7H),1.53-1.52(m,6H),1.48-1.39(br s,20H).
化合物XI-v的制备
在室温下将化合物XI-u(3.20g,4.48mmol)溶解于二氧六环中的4N HCl(50mL)中并将溶液搅拌2h。浓缩后,用MTBE洗涤残余物,得到作为灰白色固体的化合物XI-v(2.50g,92%)。
1H NMR(300MHz,CD3OD):δ7.33-7.28(m,9H),6.92-6.73(m,2H),5.12-4.99(m,4H),4.57-4.52(m,1H),3.71-3.60(m,1H),3.20-3.03(m,7H),2.81-2.53(m,8H),1.89-1.71(m,6H),1.53(br s,4H).
化合物XI-x和XI-y的制备
向化合物XI-v(2.50g,4.07mmol)和三醇XI-w(2.18g,8.14mmol)的甲醇(10mL)溶液中添加乙酸(2.5mL),并在室温下将反应混合物搅拌10分钟。添加氰基硼氢化钠(370mg,6.15mmol)并在室温下将溶液继续搅拌24h。添加额外的化合物XI-w(2.0当量)、AcOH(10当量)和NaCNBH3(1.5当量),并在室温下将该溶液继续搅拌24h。添加己醛(2.00mL,20.3mmol)、AcOH(1.10mL)和NaCNBH3(370mg,6.15mmol),并将反应混合物搅拌2h。浓缩后,使残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。分离出水层并用EtOAc(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过C18反相Gold柱纯化残余物,得到作为白色固体的化合物XI-x(310mg,7%)和化合物XI-y(510mg,14%)。
化合物XI-x的1H NMR
(400MHz,CD3OD):δ7.44(br s,5H),7.32-7.28(m,16H),6.83-6.77(m,2H),5.53-5.45(m,2H),5.18-5.04(m,4H),4.45(br s,1H),4.23-4.14(m,2H),3.97-3.91(m,6H),3.83(br s,6H),3.69(br s,2H),3.58(t,J=8.0Hz,2H),3.17-3.04(m,5H),2.76-2.51(m,12H),1.79-1.64(m,4H),1.50(br s,6H).
化合物XI-y的1H NMR
(400MHz,CD3OD):δ7.45(br s,2H),7.34-7.26(m,14H),6.79-6.75(m,2H),5.50(br s,1H),5.05(m,4H),4.55-4.44(m,1H),4.25-4.21(m,1H),3.98-3.92(m,3H),3.75-3.57(m,3H),3.20-3.29(m,6H),2.66-2.45(m,10H),1.75-1.67(m,5H),1.53(br s,6H),1.28-1.18(m,10H),0.86(t,J=7.0Hz,3H).
化合物XI-z的制备
在室温下使XI-x(310mg,0.273mmol)和10%Pd/C(30mg)在EtOH/AcOH(50mL/10mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用EtOH洗涤。将滤液在真空下浓缩,并从MTBE/己烷中沉淀,得到作为无色油状物的化合物XI-z(205mg,87%)。
1H NMR(400MHz,CD3OD):δ7.47-7.43(m,4H),7.32-7.27(m,6H),6.93-6.89(m,2H),5.52(s,2H),4.27-4.15(m,4H),3.99-3.86(m,4H),3.76-3.73(m,2H),3.16-3.09(m,3H),3.02-2.82(m,6H),2.78-2.69(m,7H),2.59(t,J=7.6Hz,2H),1.80-1.71(m,5H),1.69-1.53(m,5H).
化合物XI-bb的制备
在室温下向化合物XI-z(205mg,0.205mmol)和甲基3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(XI-aa,85mg,0.328mmol)的EtOH溶液(25mL)中添加DIPEA(211mg,1.64mmol)。将反应混合物在密封管中于70℃下加热2h,冷却至室温并在真空中浓缩。通过柱色谱(硅胶,10∶1CH2Cl2/MeOH,8∶2∶0.2CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的化合物XI-bb(160mg,63%)。
1H NMR(300MHz,CD3OD):δ7.46-7.42(m,4H),7.30-7.28(m,6H),6.89(br s,2H),5.47(s,2H),4.24-4.19(m,2H),3.99-3.83(m,6H),3.71-3.53(m,5H),2.83-2.73(m,6H),2.64-2.52(m,8H),1.77(br s,4H),1.66-1.55(m,6H).
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(化合物XI-cc)的制备:
在室温下将化合物XI-bb(160mg,0.134mmol)在4N HCl水溶液(5.0mL)中的溶液搅拌3h。除去溶剂并将通过C18反相Gold柱纯化残留物,得到作为黄色吸湿性固体的化合物XI-cc(75mg,56%)。
1H NMR(400MHz,CD3OD):δ6.99(s,2H),4.22-4.18(m,2H),3.97-3.91(m,1H),3.85-3.64(m,11H),3.50-3.35(m,10H),3.12-3.06(m,4H),2.80-2.75(m,4H),2.63(t,J=7.8Hz,2H),2.20(br,s,2H),1.81-1.64(m,8H).
方案XII
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺
化合物XII-a的制备
在室温下使XI-y(510mg,0.529mmol)和10%Pd/C(150mg)在EtOH/AcOH(50mL/10mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用EtOH洗涤。将滤液在真空下浓缩并从MTBE/己烷中沉淀,得到作为无色油状物的化合物XII-a(290mg,85%)。
1H NMR(300MHz,CD3OD):δ7.49-7.46(m,2H),7.33-7.31(m,3H),6.93(br s,2H),5.57(s,1H),4.29-4.14(m,2H),4.03-3.94(m,2H),3.81-3.57(m,2H),3.25-3.15(m,6H),3.09-2.83(m,7H),2.72-2.57(m,8H),1.80-1.55(m,12H),1.31-1.25(m,16H),0.89(t,J=6.6Hz,3H).
化合物XII-c的制备
在室温下向化合物XII-a(290mg,0.349mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(XII-b,145mg,0.560mmol)在EtOH(25mL)中的溶液中添加DIPEA(360mg,2.79mmol)。将反应混合物在密封管中于70℃下加热2h,冷却至室温并在真空中浓缩。通过柱色谱法(硅胶,10∶1CH2Cl2/MeOH,8∶2∶0.2CHCl3/CH3OH/NH4OH)纯化残余物,得到化合物为黄色固体的XII-c(250mg,65%)。
1H NMR(300MHz,CD3OD):δ7.45-7.44(m,2H),7.30-7.28(m,3H),6.91(br s,2H),5.51(s,1H),5.47(s,1H),4.26-4.21(m,1H),4.00-3.86(m,3H),3.76-3.53(m,2H),3.27-3.22(m,3H),2.93-2.67(m,7H),2.62-2.32(m,9H),1.78(br s,4H),1.67-1.50(m,6H),1.36-1.11(m,8H),0.86(t,J=6.6Hz,3H).
3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-3-氧代丙基)-5,6,7,8-四羟基萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(化合物XII-d)的盐酸盐的制备:
在室温下将化合物XII-c(250mg,0.267mmol)在4N HCl水溶液(5.0mL)中的溶液搅拌3h。除去溶剂并通过C18反相Gold柱纯化残余物,得到作为黄色吸湿性固体的化合物XII-d(125mg,55%)。
1H NMR(400MHz,CD3OD):δ6.98(s,2H),4.18-4.14(m,1H),3.84-3.76(m,2H),3.71-3.64(m,4H),3.38-3.35(m,4H),3.23-2.99(m,7H),2.80-2.75(m,4H),2.63(t,J=7.8Hz,2H),2.15-2.09(m,2H),1.81-1.64(m,10H),1.38(br,s,6H),0.93(t,J=7.0Hz,3H).
2,6-萘基酪氨酸衍生物的制备
方案XIII
3,5-二氨基-N-(N-(4-(6-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)-萘-2-基)丁基)甲脒基)6-氯吡嗪-2-甲酰胺
化合物XIII-c的制备
在室温下向化合物XIII-a(10.0g,42.1)的无水CH3CN(200mL)溶液中添加TEA(17.0g,168.7mmol)、在己烷中的10%(t-Bu)3P(1.70g,8.42mmol)、丁基-3-炔-1-醇(XIII-b,4.42g,63.1mmol)和CuI(400mg,2.10mmol)。将所得混合物用氩气脱气3分钟,一次性快速添加Pd(PPH3)4(4.86g,4.21mmol)。用氩气脱气5分钟后,将所得混合物回流4h。将反应混合物在真空下浓缩,通过柱(硅胶,80∶20己烷/EA)纯化残余物,得到作为黄色固体的化合物XIII-c(7.20g,76%)。
1H NMR(300MHz,CDCl3):δ7.85(br s,1H),7.68-7.63(m,2H),7.44-7.40(m,1H),7.16-7.08(m,2H),3.87-3.81(m,2H),3.91(s,3H),2.73(t,J=6.2Hz,2H),1.85(t,J=6.2Hz,2H).
化合物XIII-d制备
在室温下使化合物XIII-c(7.20g,31.7mmol)和10%Pd/C(2.16g)在EtOH(50mL)/AcOH(10mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用EtOH洗涤。将滤液在真空下浓缩、用饱和Na2CO3碱化,并用乙酸乙酯萃取。用水和盐水洗涤有机层,并且将有机相在减压下浓缩,得到作为黄色固体的XIII-d(5.20g,71%),其直接用于接下来的步骤。1H NMR和LC-MS数据与产物相符。
1H NMR(300
MHz,CDCl3):δ7.65(d,J=8.4Hz,2H),7.53(br s,1H),7.30-7.25(m,2H),7.12-7.10(m,2H),3.90(s,3H),3.67(t,J=6.4Hz,2H),2.77(t,J=7.2Hz,2H),1.82-1.70(m,2H),1.68-1.58(m,2H).
化合物XIII-e的制备
在室温下向化合物XIII-d(5.20g,22.5mmol)在丙酮(100mL)中的搅拌溶液中逐滴添加新制备的琼斯试剂(Jones reagent,1.3当量)。在室温下将反应混合物再搅拌30分钟,并添加琼斯试剂(0.5当量)以完成反应。将丙酮从反应混合物中倒出,用过量的丙酮洗涤固体铬盐。将丙酮层合并,用IPA淬灭,并在减压下浓缩以得到粗制品固体。该固体经酸/碱处理纯化,得到作为灰白色固体的纯的化合物XIII-e(4.20g,76%)。1H NMR和LC-MS数据与产物相符。
1H NMR(300MHz,DMSO-d6):δ12.04(s,1H),7.76-7.72(m,2H),7.59(br s,1H),7.33-7.26(m,2H),7.14-7.10(m,1H),3.85(s,3H),2.71(t,J=7.4Hz,2H),2.24(t,J=7.4Hz,2H),1.92-1.82(m,2H).
化合物XIII-g的制备
向化合物XIII-e的无水THF(50mL)溶液(4.20g,17.1mmol)中添加三乙胺(4.30g,42.8mmol)和新戊酰氯(2.46g,20.5mmol),然后加入氯化锂(860mg,20.5mmol)。在-25℃添加化合物XIII-f(3.6g,20.5mmol),在室温下将反应混合物搅拌2h。反应完成后,将反应混合物蒸发并用IN NaOH研磨残余物。分离出水层并用二氯甲烷(2×500mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过MTBE和己烷洗涤纯化残余物,得到作为灰白色固体的化合物XIII-g(5.50g,79%)。1H NMR和LC-MS数据与产物相符。
1H NMR(400MHz.CDCl3):δ7.77-7.63(m,2H),7.58(br s,1H),7.33-7.23(m,5H),7.18-7.16(m,2H),7.13-7.10(m,2H),4.62-4.56(m,1H),4.12-4.07(m,2H),3.26-3.22(m,1H),3.08-2.94(m,2H),2.86-2.82(m,2H),2.72-2.66(m,1H),2.15-2.07(m,2H).
化合物XIII-h的制备
在-78℃向化合物XIII-g(11.2g,27.79mmol)的无水THF(300mL)溶液中分批添加KHMDS(7.18g,36.1mmol)。将所得的混合物搅拌30分钟,添加三异丙基苯磺酰叠氮化物(12.8g,41.68mmol)并将反应混合物搅拌2-3分钟。在相同的温度下缓慢添加乙酸(10.0g,166.7mmol),随后添加乙酸四甲基铵(14.7g,111.1mmol)。将混合物升温至27℃,搅拌4h,并用饱和NaHCO3(300mL)淬灭,浓缩至除去THF,并且用EtOAc(2×500mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过柱色谱(硅胶,70∶30己烷/EtOAc)纯化残余物,得到作为无色油状物的化合物XIII-h(8.10g,65%),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-i的制备
在0℃向化合物XIII-h在THF/H2O(100mL/25mL)中的溶液(8.10g,18.2mmol)中添加H2O2(3.7g,109.2mmol),随后分批添加LiOH(1.56g,36.4mmol)。在相同的温度下将反应混合物搅拌1h,用饱和Na2SO3(200mL)淬灭,在减压下浓缩以除去THF并用CH2Cl2(200mL)洗涤。用1N HCl水溶液使水层酸化并用CH2Cl2(2×250mL)萃取。将合并的有机萃取物经Na2SO4干燥、浓缩并用MTBE洗涤,得到作为灰白色固体的化合物XIII-i(4.10g,80%),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-j的制备
在室温下使化合物XIII-i(4.10g,14.3mmol)和10%Pd/C(410mg)在AcOH/H2O(50mL/15mL)中的混悬液置于氢化条件(1atm)下3h。将反应混合物通过硅藻土过滤并用MeOH洗涤。将滤液在真空下浓缩,得到作为白色固体的乙酸盐XIII-j(3.40g,91%),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-k的制备
在室温下向化合物XIII-j(3.40g,13.1mmol)的乙酸(30mL)溶液中逐滴添加氢溴酸(30mL),并将反应混合物回流4h。将反应混合物冷却至室温并浓缩。在减压下浓缩残余物,得到作为棕色固体的化合物XIII-k(2.60g,81%)。1H NMR和LC-MS数据与产物相符。
1H NMR and LC-MS data is consistent with product.1H NMR(400MHz,CD3OD):δ7.72-7.59(m,3H),7.45-7.27(m,1H),7.19-7.03(m,1H),4.01(t,J=5.8Hz,1H),2.99-2.81(m,2H),2.38-2.14(m,2H).
化合物XIII-l的制备
在0℃向化合物XIII-k(13.8g,56.3mmol)在MeOH/H2O(160mL/100mL)的溶液中添加NaHCO3(4.50g,112.6mmol)和Boc2O(14.7g,67.5mmol)。将所得混合物升温至室温并搅拌3h。使反应混合物在CH2Cl2(200mL)与水(200mL)之间分配。分离出水层并用CH2Cl2(2×200mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为白色固体的化合物XIII-l(14.0g,73%)。1H NMR和LC-MS数据与产物相符。
1H NMR(400MHz,DMSO-d6):δ12.43(br s,1H),9.58(br s,1H),7.66-7.58(m,2H),7.52(br s,1H),7.26-7.21(m,2H),7.07-7.02(m,2H),3.88-3.82(m,1H),2.79-2.64(m,2H),2.01-1.86(m,2H),1.40(s,9H).
化合物XIII-m的制备
在0℃向酸XIII-l(13.7g,39.7mmol)在THF(150mL)中的溶液中添加DIPEA(7.68g,59.5mmol)和T3P(18.9g,59.5mmol)。在相同温度下将反应混合物搅拌1h,逐滴添加NH3(在甲醇中7.0N,29.4mL,206mmol)。在0℃下将反应混合物搅拌1h,升温至室温并搅拌1h。浓缩后,使残余物在CH2Cl2(100mL)与水(100mL)之间分配。分离出水层并用CH2Cl2(2×100mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。将残余物用MTBE洗涤,得到作为浅黄色固体的酰胺XIII-m(7.20g,53%)。1H NMR和LC-MS数据与产物相符。
1H NMR和LC-MS数据与产物相符。1H NMR(300MHz,DMSO-d6):δ9.60(s,1H),7.66-7.51(m,3H),7.28-7.16(m,4H),7.06-6.94(m,4H),6.15(br s,1H),3.90-3.82(m,1H),2.84-2.58(m,2H),1.98-1.80(m,2H),1.40(s,9H).
化合物XIII-n的制备
在0℃向化合物XIII-m(7.20g,20.9mmol)吡啶(70mL)溶液中添加三氟甲磺酸酯(8.90g,31.3mmol),在室温下将反应混合物搅拌1h。浓缩后,使反应混合物在CH2Cl2(300mL)与水(300mL)之间分配。分离出水层并用CH2Cl2(2×300mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为棕色固体的化合物XIII-n(6.80g,69%),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-p的制备
在室温下向化合物XIII-n(6.80g,14.2)的无水CH3CN(150mL)溶液中添加TEA(5.7g,57.1mmol)、在己烷中的10%(t-Bu)3P(0.57g,2.84mmol)、丁-3-炔基氨基甲酸苄酯(XIII-o,4.30g,21.5mmol),和CuI(134mg,0.71mmol)。将所得混合物用氩气脱气3分钟,一次性迅速添加Pd(PPH3)4(1.60g,1.42mmol)。用氩气脱气5分钟后,将所得混合物回流4h。将反应混合物在真空下浓缩,通过柱(硅胶,80∶20己烷/EA)纯化残余物,得到作为棕色固体的化合物XIII-p(4.50g,60%)。
1H NMR(300MHz,CD3OD):δ7.83(s,1H),7.72-7.60(m,3H),7.43-7.24(m,7H),5.10(s,2H),4.04(br s,1H),3.37(t,J=6.8Hz,2H),2.85-2.75(m,2H),2.64(t,J=6.8Hz,2H),2.20-1.90(m,2H),1.45(s,9H).
化合物XIII-q的制备
在室温下使化合物XIII-p(4.50g,8.50mmol)和10%Pd/C(135mg)在EtOH(500mL)/AcOH(10mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤,并用EtOH洗涤。将滤液在真空下浓缩,并用MTBE/己烷洗涤,得到作为灰白色固体的乙酸盐(4.20g,粗制品),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-q的制备
在0℃向粗制化合物XIII-q(4.20g,粗制品)在MeOH/H2O(100mL/500mL)的搅拌溶液中添加饱和Na2CO3和CbzCl(2.68g,15.7mmol),并在相同温度下搅拌1h。在室温下将反应混合物搅拌1h。除去溶剂,使混合物在CH2Cl2(500mL)与水(100mL)之间分配。分离出水层并用CH2Cl2(2×100mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为黄色油状物的化合物XIII-q(2.80g,经两步的产率62%),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-r的制备
向化合物XIII-q(2.80g,5.25mmol)的二氧六环溶液中添加二氧六环中的4N HCl(30mL),并在室温下将反应混合物搅拌4h。在真空下除去溶剂,并将残余物用MTBE洗涤,得到化合物XIII-r(1.90g,82%)。1H NMR和LC-MS数据与产物相符。
1H NMR(400MHz,CD3OD):δ7.75-7.69(m,2H),7.64-7.54(m,2H),7.36-7.25(m,8H),5.04(s,2H),3.98(t,J=6.4Hz,1H),3.14(t,J=6.8Hz,2H),2.87(t,J=7.8Hz,2H),2.77(t,J=7.4Hz,2H),2.33-2.15(m,2H),1.75-1.68(m,2H),1.58-1.51(m,2H).
化合物XIII-s的制备
向化合物XIII-r(1.90g,4.38mmol)和醛2(910g,5.26mmol)的MeOH(80mL)溶液中添加乙酸(2.6g,43.8mmol),在室温下将反应混合物搅拌10分钟。添加氰基硼氢化钠(413mg,6.57mmol)并在室温下将溶液继续搅拌1h。添加额外的化合物2(0.3当量)、AcOH(0.5当量)和NaCNBH3(0.5当量)并搅拌1h。浓缩后,使残余物在EtOAc(300mL)和饱和NaHCO3(200mL)之间分配。分离出水层并用EtOAc(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩,得到粗制残余物XIII-S(2.50g),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-t的制备
在0℃向化合物XIII-s(2.50g,在MeOH/H2O(80mL/30mL)的粗制品)中添加饱和Na2CO3,将溶液搅拌10分钟。逐滴添加氯甲酸苄酯(1.0g,6.30mmol),并在0℃下将反应混合物搅拌1h,升温至室温并搅拌1h。浓缩后,残余物溶解于CH2Cl2(200mL)中,然后用水(300mL)和盐水(300mL)洗涤。将有机层经Na2SO4干燥、浓缩并通过柱色谱纯化,得到XIII-t(1.90g,经两步的产率62%),其直接用于接下来的步骤。LC-MS数据与产物相符。
化合物XIII-u的制备
在室温下将化合物XIII-t(1.90g,2.62mmol)溶解于二氧六环中的4N HCl(30mL)中,将溶液搅拌2h。浓缩后,用MTBE洗涤残余物,得到作为灰白色固体的化合物XIII-u(1.30g,82%)。1H NMR和LC-MS数据与产物相符。
1H NMR(300MHz,CD3OD):δ7.69-7.63(m,2H),7.56-7.55(m,2H),7.35-7.21(m,16H),5.15-5.04(m,5H),4.59-4.45(m,2H),3.75-3.59(m,9H),3.57-3.34(m,3H),3.20-3.12(m,5H),3.08-2.94(m,6H),2.76(t,J=7.4Hz,4H),2.35-2.15(m,3H),2.02-1.81(m,6H),1.76-1.49(m,4H).
化合物XIII-w和XIII-x的制备
向化合物XIII-u(1.30mg,2.08mmol)和三醇XIII-v(1.08g,4.16mmol)的甲醇(80mL)溶液中添加乙酸(1.2g,20.8mmol),在室温下将反应混合物搅拌10分钟。添加氰基硼氢化钠(193mg,3.12mmol),并在室温下将溶液继续搅拌24h。添加额外的化合物XIII-x(2.0当量),AcOH(4.0当量)和NaCNBH3(3.0当量),并在室温下将该溶液继续搅拌24h。添加己醛(1.0mL,10.4mmol)、AcOH(1.10mL)和NaCNBH3(193mg,3.12mmol),并将反应混合物搅拌2h。浓缩后,使残余物在EtOAc(300mL)和饱和NaHCO3(200mL)之间分配。分离出水层并用EtOAc(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过C18反相Gold柱纯化残余物,得到作为白色固体的化合物XIII-w(550mg,25%)和化合物XIII-x(400mg,21%)。1H NMR和LC-MS数据与产物相符。
化合物XIII-w的1H NMR
(300MHz,CD3OD):δ7.67-7.65(m,2H),7.55-7.40(m,3H),7.31-7.26(m,22H),5.42-5.29(m,2H),5.04(s,4H),4.21-4.15(m,2H),3.94-3.84(m,6H),3.68-3.50(m,5H),3.14(t,J=6.8Hz,2H),2.78-2.61(m,10H),1.73-1.68(m,4H),1.58-1.51(m,2H).
化合物XIII-x的1H NMR
(400MHz,CD3OD):δ7.68(d,J=7.4Hz,2H),7.53-7.49(m,2H),7.42-7.27(m,16H),5.48-5.42(m,1H),5.11(br s,2H),5.04(s,2H),4.50-4.38(m,1H),4.23-4.19(m,1H),3.97-3.88(m,3H),3.75-3.48(m,3H),3.13-3.10(m,3H),2.78-2.71(m,5H),2.48-2.29(m,5H),2.15-2.02(m,1H),1.80-1.51(m,6H),1.33-1.14(m,6H),0.82(t,J=6.8Hz,3H).
化合物XIII-z的制备
在室温下使XIII-y(550mg,0.487mmol)和10%Pd/C(165mg)在EtOH/AcOH(50mL/10mL)中的混悬液置于氢化条件(1atm)下8h。将反应混合物通过硅藻土过滤并用EtOH洗涤。将滤液在真空下浓缩,并从MTBE/己烷中沉淀,得到作为无色油状物的化合物XIII-z(400mg,95%)。
1H NMR(300MHz,CD3OD):δ7.73-7.56(m,4H),7.41-7.23(m,14H),5.45(s,2H),4.25-4.13(m,5H),3.98-3.83(m,5H),3.74-3.50(m,6H),3.13-2.98(m,5H),3.00-2.71(m,10H),1.82-1.66(m,7H).
化合物XIII-aa的制备
在室温下向化合物XIII-z(400mg,0.465mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(XIII-aa,193mg,0.744mmol)在EtOH(50mL)中的溶液中添加DIPEA(480mg,3.72mmol)。将反应混合物在密封管中于70℃下加热2h,冷却至室温并在真空中浓缩。通过柱色谱(硅胶,10∶1CH2Cl2/MeOH,8∶2∶0.2CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的化合物XIII-bb(350mg,70%)。
1H NMR(300MHz,CD3OD):δ7.72-7.67(m,2H),7.60(d,J=9.6Hz,2H),7.44-7.41(m,4H),7.34-7.26(m,8H),5.49(s,1H),5.45(s,2H),4.24-4.18(m,2H),3.99-3.90(m,4H),3.85-3.82(m,2H),3.71-3.68(m,2H),3.56(t,J=10.5Hz,2H),3.16-3.06(m,1H),2.85-2.74(m,4H),2.68-2.62(m,4H),2.58-2.47(m,3H),1.98-1.81(m,6H),1.70-1.58(m,4H).
3,5-二氨基-N-(N-(4-(6-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(XIII-bb)的盐酸盐的制备:
在室温下将化合物XIII-aa(350mg,0.326mmol)在IN HCl水溶液(5.0mL)中的溶液搅拌3h。除去溶剂并且通过C18反相Gold柱纯化残留物,得到作为黄色吸湿性固体的化合物XIII-bb(250mg,86%)。
1H NMR(400MHz,CD3OD)∶δ7.75(d,J=7.8Hz,2H),7.60(br,s,2H),7.37-7.32(m,2H),4.16(br,s,2H),3.81-3.75(m,4H),3.73-3.63(m,6H),3.48-3.44(m,2H),3.37-3.34(m,7H),3.13-3.10(m,H),2.85(t,J=2Hz,4H),2.25-2.15(m,3H),1.92-1.69(m,4H).
方案XIV
3,5-二氨基-N-(N-(4-(6-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺
化合物XIV-a的制备
在室温下使XIII-x(400mg,0.416mmol)和I0%Pd/C(120mg)在EtOH/AcOH(50mL/10mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用MeOH洗涤。将滤液在真空下浓缩,得到作为无色油状物的化合物XIV-a(270mg,93%)。
1H NMR(400MHz,CD3OD):δ7.73(d,J=8.2Hz,2H),7.62(d,J=6.2Hz,2H),7.48-7.39(m,3H),7.36-7.24(m,6H),5.52(s,1H),4.23-4.15(m,2H),4.00-3.94(m,2H),3.80-3.77(m,1H),3.64-3.57(m,2H),3.22-3.07(m,4H),3.04-2.91(m,3H),2.85-2.75(m,6H),1.82-1.77(m,4H),1.73-1.54(m,5H),1.33-1.11(m,10H),0.82(t,J=6.8Hz,3H).
化合物XIV-c的制备
在室温下向化合物XIV-a(270mg,0.390mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(XIV-b,163mg,0.62mmol)的EtOH(50mL)溶液中添加DIPEA(402mg,3.12mmol)。将反应混合物在密封管中于70℃下加热2h,冷却至室温并在真空中浓缩。通过柱色谱法(硅胶,10∶1 CH2Cl2/MeOH,8∶2∶0.2 CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的化合物XIV-c(180mg,57%)。
1H NMR(400MHz,CD3OD):δ7.71-7.68(m,2H),7.60-7.59(m,2H),7.46-7.42(m,2H),7.34-7.25(m,5H),5.50(s,1H),4.25-4.21(m,1H),4.01-3.88(m,3H),3.76-3.71(m,1H),3.63-3.55(m,1H),3.15-3.09(m,1H),2.84-2.73(m,5H),2.61-2.43(m,8H),2.02-1.79(m,4H),1.73-1.59(m,4H),1.43-1.41(m,2H),1.29-1.15(m,8H),2.02-1.79(m,4H),1.73-1.59(m,4H),1.43-1.41(m,2H),1.29-1.15(m,8H),0.84(t,J=6.8Hz,3H).
3,5-二氨基-N-(N-(4-(6-((S)-4-氨基-3-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)萘-2-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(XIV-d)的制备
在室温下将化合物XIV-c(180mg,0.199mmol)在4N HCl水溶液(2.0mL)中的溶液搅拌2h。除去溶剂并且通过C18反相Gold柱纯化残留物,得到作为黄色吸湿性固体的化合物XIV-d(82mg,50%)。
1H NMR(400MHz,CD3OD):δ7.75(d,J=8.4,Hz,2H),7.66(d,J=8.4Hz,2H),7.38(t,J=7.8Hz,2H),4.18-4.16(m,1H),4.04-4.02(m,1H),3.85-3.76(m,2H),3.71-3.64(m,3H),3.48-3.46(m,1H),3.38-3.34(m,8H),3.25-3.08(m,5H),2.92-2.81(m,4H),1.38(br s,6H),0.93(t,J=6.6Hz,3H).
方案XV
3,5--二氨基-N-(N-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)-5,6,7,8-四羟基萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(XV-dd):
化合物XV-b的制备
在0℃向化合物XV-a(100g,674mmol)的无水THF溶液(600mL)中添加硫酸二甲酯(102g,809mmol),随后添加NaOH(32.4g,809mmol)。在室温下将所得反应混合物搅拌3h。反应完成后,将反应混合物浓缩以除去溶剂并用水稀释。用乙酸乙酯(2×500mL)萃取水层。将合并的有机萃取物经Na2SO4干燥并浓缩。通过柱色谱法(硅胶,90∶10己烷/EtOAc)纯化残余物,得到作为无色油状物的化合物XV-b(108g,98%)。
1H NMR(400MHz,CDCl3):δ7.05(t,J=7.8Hz,1H),6.69(d,J=7.6Hz,1H),6.64(d,J=7.9Hz,1H),3.80(s,3H),2.74(t,J=6.2Hz,2H),2.64(t,J=6.2Hz,2H),1.81-1.71(m,4H).
化合物XV-c的制备
在0℃向琥珀酸酐(12.3g,123mmol)的CH2Cl2(150mL)溶液中分批添加AlCl3(18.4g,138mmol)。10分钟后,在相同的温度下将溶于CH2Cl2(50mL)的化合物XV-b(20.0g,123mmol)添加到反应混合物中。在室温下将反应混合物搅拌3h。反应完成后,将反应混合物倒入冰冷的水中并用HCl酸化。将反应混合物通过硅藻土垫过滤以除去Al(OH)3并用热乙酸乙酯洗涤。用乙酸乙酯萃取水层。将溶剂浓缩以得到固体,通过用己烷洗涤研磨该化合物使得其进一步纯化以得到作为白色固体的化合物XV-c(22.3g,69%)。1H NMR和LC-MS数据与产物相符。
1H NMR(400MHz,CD3OD):δ7.68(d,J=8.7Hz,1H),6.80(d,J=8.7Hz,1H),3.86(s,3H),3.16(t,J=6.4Hz,2H),2.90(t,J=6.4Hz,2H),2.67-2.60(m,4H),1.78-1.64(m,4H).
化合物XV-d的制备
在室温下向化合物XV-c(30g,114mmol)在甲苯(300mL)中的溶液中添加浓盐酸(300mL),随后分批添加Zn粉(74.8g,1145mmol)。将反应混合物加热至回流3h,冷却至室温并通过硅藻土过滤。在滤液浓缩至50%后,将所得的沉淀物过滤并干燥,得到作为灰白色固体的化合物XV-d(24.0g,85%)。
1H NMR(400MHz,CD3OD):δ6.90(d,J=8.4Hz,1H),6.63(d,J=8.4Hz,1H),3.75(s,3H),2.67(t,J=5.9Hz,2H),2.61(t,J=5.9Hz,2H),2.57-2.49(m,2H),2.35-2.28(m,2H),1.85-1.68(m,4H).
化合物XV-e的制备
在-25℃向化合物XV-d(20g,80.6mmol)的无水THF(500mL)溶液中添加Et3N(28mL,96.8mmol)、PivCl(11.9mL,96.7mmol)和LiCl(3.418g,96.8mmol),随后添加化合物XV-f(17.1g,96.8mmol),在室温下将反应混合物搅拌2h。反应完成后,将反应混合物蒸发,用1N NaOH处理残余物。分离出水层并用二氯甲烷(2×500mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过用MTBE和己烷研磨来纯化残余物以得到作为固体的化合物XV-e(26g,79%)。
1H NMR(400MHz,CDCl3):δ7.37-7.27(m,3H),7.23-7.15(m,2H),6.98(d,J=8.5Hz,1H),6.63(d,J=8.5Hz,1H),4.70-4.61(m,1H),4.21-4.11(m,2H),3.78(s,3H),3.30(dd,J=9.7Hz,1H),3.14-2.79(m,3H),2.80-2.52(m,7H),2.05-1.88(m,2H),1.89-1.64(m,4H).
化合物XV-g的制备
在-78℃向化合物XV-e的无水THF(300mL)溶液(30.0mL g,73.7mmol)中分批添加KHMDS(19.1g,95.8mmol)。在将所得混合物搅拌30分钟后,添加三异丙基苯磺酰叠氮化物(34.2g,110.6mmol)并将反应混合物搅拌2-3分钟。在相同温度下缓慢添加乙酸(26.5g,442mmol),随后添加乙酸四甲基铵(29.5g,221mmol)。将反应混合物升温至27℃,搅拌4h,用饱和NaHCO3(300mL)淬灭,浓缩以除去THF并且用EtOAc(2×500mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过柱色谱法(硅胶,70∶30己烷/EtOAc)纯化残余物,得到作为无色油状物的化合物XV-g(18.3g,55%)。
1H NMR(400MHz,CDCl3):δ7.37-7.27(m,3H),7.23-7.18(m,2H),6.96(d,J=8.4Hz,1H),6.62(d,J=8.4Hz,1H),5.03-4.98(m,1H),4.61-4.54(m,2H),4.23-4.10(m,2H),3.77(s,3H),3.32(dd,J=10.3Hz,1H),2.86-2.77(m,2H),2.73-2.61(m,5H),2.17-2.07(m,1H),2.05-1.95(m,1H),1.82-1.70(m,4H).
化合物XV-h的制备
在0℃向化合物XV-g(40.5g,20.0mmol)的THF/H2O(150mL/50mL)溶液中添加H2O2(61.4mL,542mmol),随后分批添加LiOH(7.57g,181mmol)。在相同温度下将反应混合物搅拌1h,用饱和Na2SO3水溶液(200mL)淬灭,在减压下浓缩以除去THF并用CH2Cl2(200mL)洗涤。用1N HCl水溶液使水层酸化并用CH2Cl2(2×250mL)萃取。将合并的有机萃取物经Na2SO4干燥、浓缩并用MTBE研磨,得到作为灰白色固体的化合物XV-h(20.0g,77%)。
1H NMR(300MHz,CD3OD):δ6.91(d,J=8.4Hz,1H),6.64(d,J=8.4Hz,1H),3.93(dd,J=7.9,4.1Hz,1H),3.74(s,3H),2.72-2.55(m,6H),2.08-1.83(m,2H),1.81-1.63(m,4H).
化合物XV-i的制备
在室温下使化合物XV-h(41.0g,144mmol)和10%Pd/C(8.0g)在AcOH/H2O(480mL/160mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用MeOH洗涤。将滤液在真空下浓缩,得到作为黄色固体的乙酸盐XV-i(40.0g,86%)。
1H NMR(400MHz,DMSO-d6):δ6.98(d,J=8.8Hz,1H),6.69(d,J=8.8Hz,1H),3.97(t,J=6.3Hz,1H),3.76(s,3H),2.76-2.55(m,7H),2.14-1.99(m,2H),1.84-1.67(m,5H),1.93(s,3H).
化合物XV-j的制备
在室温下向化合物XV-i(41.3g,128mmol)的乙酸(250mL)溶液中逐滴添加氢溴酸(250mL),将反应混合物回流16h。将反应混合物冷却至室温并浓缩。用H2O(15mL)稀释残余物,用氨水使残余物略微碱化,并过夜结晶,得到作为棕色固体的化合物XV-j(40.0g,95%)。
1H NMR(400MHz,DMSO-d6):δ8.35(brs,1H),6.74(d,J=7.7Hz,1H),6.56(d,J=7.7Hz,1H),4.01-3.90(m,1H),2.55-2.38(m,4H),2.05-1.85(m,2H),1.78-1.56(m,6H),
化合物XV-k的制备
在0℃将乙酰氯(60.5mL,852mmol)添加到无水甲醇(400mL),中,随后添加化合物XV-j(40.0g,122mmol)。将反应混合物回流3h并浓缩。使残余物在CH2Cl2(500mL)与饱和NaHCO3(300mL)之间分配。分离出水层并用CH2Cl2(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩,得到作为无色油状物的化合物XV-k(30.0g,83%)。
1H NMR(400MHz,CD3OD):δ6.77(d,J=8.2Hz,1H),6.50(d,J=8.2Hz,1H),3.72(s,3H),3.51(t,J=6.2Hz,1H),2.70-2.57(m,4H),2.53(t,J=8.5Hz,2H),1.83-1.69(m,6H).
化合物XV-l的制备
在0℃向化合物XV-k(30.0g,114mmol)的MeOH/H2O溶液(300mL/100mL)中添加NaHCO3(39.0g,456mmol)和Boc2O(30.0g,137mmol)。将所得混合物升温至室温并搅拌3h。使反应混合物在CH2Cl2(200mL)与水(200mL)之间分配。分离出水层并用CH2Cl2(2×200mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥,并通过柱色谱(硅胶,70∶30己烷/EA)纯化残余物,得到作为无色油状物的化合物XV-l(31.0g,85%)。
1H NMR(400MHz,CDCl3):δ6.80(d,J=8.0Hz,1H),6.56(d,J=8.0Hz,1H),5.32(brs,1H),5.29(s,1H),4.45-4.30(m,1H),3.73(s,3H),2.69-2.43(m,6H),1.88-1.72(m,6H),1.46(s,9H).
化合物XV-m的制备
在0℃向化合物XV-l(31.0g,85.4mmol)的吡啶(70mL)溶液中添加三氟甲磺酸酯(21.5mL,128mmol),并在室温下将反应混合物搅拌1h。浓缩后,使反应混合物在CH2Cl2(300mL)与水(300mL)之间分配。分离出水层并用CH2Cl2(2×300mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为棕色油状物的化合物XV-m(41.0g粗制品)。
1H NMR(400MHz,CDCl3):δ7.01(s,2H),5.22-5.11(m,1H),4.44-4.34(m,1H),3.74(s,3H),2.78(t,J=6.0Hz,2H),2.71-2.52(m,4H),2.16-2.02(m,1H),1.89-1.73(m,5H),1.45(s,9H).
化合物XV-o的制备
在室温下向化合物XV-m(41.0g,粗制品)的无水CH3CN(400mL)溶液中添加TEA(46.8mL,342mmol)、在己烷中的10%(t-Bu)3P(34.5mL,17.0mmol)、丁-3-炔基氨基甲酸苄酯(XV-n,20.6g,103mmol)和CuI(0.81g,4.26mmol)。将所得混合物用氩气脱气3分钟,一次性快速添加Pd(PPH3)4(9.86g,8.53mmol)。用氩气脱气5分钟后,将所得混合物回流16h。将反应混合物在真空下浓缩,通过柱(硅胶,80∶20己烷/EA)纯化残余物,得到作为棕色油状物的化合物XV-o(25g,经两步的产率54%)。
1H NMR(400MHz,CDCl3):δ7.38-7.31(m,5H),7.15(d,J=7.9Hz,1H),6.88(d,J=7.9Hz,1H),5.19-5.05(m,4H),4.43-4.31(m,1H),3.73(s,3H),3.47-3.37(m,2H),2.82(t,J=6.7Hz,2H),2.69-2.55(m,5H),2.13-2.00(m,1H),1.82-1.70(m,6H),1.45(s,9H).
化合物XV-p的制备
向甲酯XV-o(23.0g,42.0mmol)的THF/MeOH/H2O(200mL/200mL/65mL)溶液中添加NaOH(10.0g,252mmol),并在室温下将反应混合物搅拌温度1h。用1N HCl水溶液将pH值调节至9并除去该有机溶剂。将残余物的pH值调节至5,并使该混悬液在CH2Cl2(200mL)与水(200mL)之间分配。分离出水层并用CH2Cl2(2×200mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩,得到作为棕色固体的化合物XV-p(16.0g,72%)。
1H NMR(400MHz,CD3OD):δ7.38-7.20(m,5H),7.09(d,J=8.6Hz,1H),6.89(d,J=8.5Hz,1H),5.08(s,2H),4.16-4.02(m,1H),3.34(t,J=7.3Hz,2H),2.81(t,J=6.4Hz,2H),2.70-2.58(m,6H),2.08-1.93(m,1H),1.90-1.81(m,1H),1.80-1.68(m,4H),1.45(s,9H).
化合物XV-q的制备
在0℃向酸XV-p(11.0g,20.6mmol)的THF(200mL)溶液中添加NMM(3.39mL,31.0mmol)和PivCl(3.0mL,24.7mmol)。在相同温度下将反应混合物搅拌1h并逐滴添加NH3(在甲醇中7.0N,29.4mL,206mmol)。在0℃下将反应混合物继续搅拌1h,升温至室温并搅拌1h。浓缩后,使残余物在CH2Cl2(100mL)与水(100mL)之间分配。分离出水层并用CH2Cl2(2×100mL)萃取。将混合有机萃取物经Na2SO4干燥并浓缩。用MTBE研磨残余物,得到作为黄色固体的酰胺XV-q(12.0g,粗制品)。
1H NMR(400MHz,CD3OD):δ7.35-7.22(m,5H),7.09(d,J=8.3Hz,1H),6.88(d,J=8.3Hz,1H),5.07(s,2H),4.09-3.97(m,1H),3.39(t,J=6.9Hz,2H),2.80(t,J=6.5Hz,2H),2.70-2.56(m,6H),1.82-1.67(m,6H),1.45(s,9H).
化合物XV-r的制备
在室温下使化合物XV-q(12.0g,粗制品)和10%Pd/C(2.50g)在EtOH(300mL)/AcOH 960mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用EtOH洗涤。将滤液在真空下浓缩并用MTBE/己烷研磨,得到作为灰白色固体的乙酸盐XV-r(12.0g,粗制品)。该产物直接用于接下来的步骤。[M+H]+264。
化合物XV-s的制备
在0℃下向化合物XV-r(12.0g,粗制品)在MeOH(300mL)/水中的搅拌溶液(100mL)中添加Na2CO3(21.8g,206mmol)和CbzCl(6.27mL,41.2mmol)并在相同温度下搅拌1h。在室温下将反应混合物搅拌1h,除去溶剂,混合物在CH2Cl2(500mL)与水(100mL)之间分配。分离出水层并用CH2Cl2(2×100mL)萃取。将合并的有机萃取物用盐水洗涤,经Na2SO4干燥并浓缩,得到作为黄色油状物的化合物XV-s(15.0g,粗制品)。该产物直接用于接下来的步骤。[M+H]+538。
化合物XV-t的制备
向化合物XV-s(15.0g,粗制品)的溶液中添加二氧六环中的4N HCl溶液(60mL),并在室温下将反应混合物搅拌1h。在真空下除去溶剂并用MTBE研磨残余物,得到化合物XV-t(15.0g,粗制品)。该产物直接用于接下来的步骤。[M+H]+438。
化合物XV-u的制备
向化合物XV-t(15.0g,粗制品)和醛2(4.27g,24.2mmol)的MeOH(100L)溶液中添加乙酸(12.5mL),并在室温下将反应混合物搅拌10分钟。添加氰基硼氢化钠(1.94g,30.9mmol),并在室温下将该溶液继续搅拌1h。添加额外的化合物XV-u(0.3当量)、AcOH(0.5当量)和NaCNBH3(0.5当量),搅拌1h。浓缩后,使残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。分离出水层并用EtOAc(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。将残余物(20g,粗制品)无需进一步纯化直接用于接下来的步骤。[M+H]+595。
化合物XV-w的制备
在0℃下向化合物XV-u(20g,在MeOH/H2O(300mL/100mL)中的粗制品)的溶液中添加Na2CO3(21.8g,206mmol)并将该溶液搅拌10分钟。逐滴添加氯甲酸苄酯(6.77mL,41.2mmol),并在0℃下将反应混合物搅拌1h,升温至室温并搅拌1h。浓缩后,残余物溶于CH2Cl2(200mL)中,并用水(300mL)和盐水(300mL)洗涤。将有机层经Na2SO4干燥并浓缩。将将残余物(20g,粗制品)无需进一步纯化直接用于接下来的步骤。[M+Na]+752。
化合物XV-w的制备
在室温下将化合物XV-v(20g,粗制品)溶解于二氧六环中的4N HCl(50mL)中并将溶液搅拌2h。浓缩后,用MTBE研磨残余物,用NaHCO3水溶液中和,并使用CMA系统通过快速柱色谱法纯化得到作为灰白色固体的化合物XV-w(3.50g,经7步产率27%)。
1H NMR(400MHz,CD3OD):δ7.43-7.24(m,10H),6.83(s,2H),5.16(s,2H),5.05(s,2H),4.54-4.42(m,1H),3.58-3.44(m,1H),3.42-3.33(m,2H),3.15-3.10(m,2H),3.00-2.86(m,2H),2.71-2.57(m,4H),2.56-2.45(m,5H),2.32-1.87(m,2H),1.79-1.65(m,4H),1.59-1.48(m,4H).
化合物XV-y和XV-z的制备
向化合物XV-w(3.50mg,5.57mmol)和三醇XV-x(6.00g,22.3mmol)在甲醇(50mL)中的溶液中添加乙酸(3.35mL),将在室温下反应混合物中的溶液搅拌10分钟。添加氰基硼氢化钠(1.40g,22.3mmol),并在室温下将该溶液继续搅拌24h。添加额外的化合物XV-x(2.0当量)、AcOH(4.0当量)和NaCNBH3(3.0当量),并在室温下将该溶液继续搅拌24h。添加己醛(2.00mL,16.8mmol)、AcOH(1.10mL)和NaCNBH3(1.75g,27.9mmol),并将反应混合物搅拌2h。浓缩后,使残余物在EtOAc(300mL)与饱和NaHCO3(200mL)之间分配。分离出水层并用EtOAc(2×300mL)萃取。将合并的有机萃取物经Na2SO4干燥并浓缩。通过C18反相Gold柱纯化残余物,得到作为白色固体的化合物XV-z(2.50g,40%)和化合物XV-y(1.30g,24%):
1H NMR(400MHz,CD3OD):δ7.51-7.19(m,20H),6.87-6.74(m,2H),5.51-5.32(m,2H),5.16-5.20(m,2H),4.45-4.25(m,1H),4.20(dd,J=10.8,5.6Hz,2H),4.00-3.90(m,3H),3.89-3.80(m,2H),3.75-3.63(m,2H),3.55(t,J=11.3Hz,2H),3.11(t,J=6.5Hz,2H),2.80-2.36(m,12H),2.20-2.01(m,1H),1.99-1.83(m,1H),1.82-1.62(m,6H),1.57-1.44(m,4H),1.41-1.21(m,2H),0.94-0.86(m,1H).
化合物XV-aa的制备
在室温下使XV-z(2.50g,2.20mmol)和10%Pd/C(500mg)在EtOH/AcOH(100mL/20mL)中的混悬液置于氢化条件(1atm)下16h。将反应混合物通过硅藻土过滤并用MeOH洗涤。将滤液在真空下浓缩并用MTBE/己烷研磨,得到作为白色固体的化合物XV-aa(2.20g,96%)。
1H NMR(400MHz,CD3OD):δ7.45-7.38(m,4H),7.30-7.24(m,6H),6.87(s,2H),5.47(s,2H),4.23(dd,J=10.9,5.7Hz,2H),4.17-4.10(m,2H),3.98-3.90(m,2H),3.84(dd,J=5.1,2.3Hz,2H),3.72(dd,J=9.4,2.3Hz,2H),3.65-3.54(m,4H),3.13-2.96(m,4H),2.93-2.81(m,2H),2.80-2.47(m,11H),.95(s,9H),1.81-1.54(m,10H),1.40-1.23(m,2H).
化合物XV-cc的制备
在室温下向化合物XV-aa(2.20g,2.10mmol)和3,5-二氨基-6-氯吡嗪-2-羰基甲脒基硫代酸甲酯氢碘酸盐(XV-bb,1.30g,3.36mmol)的EtOH(15mL)溶液中添加DIPEA(2.98mL,16.8mmol)。将反应混合物在密封管中于70℃下加热2h,冷却至室温并在真空中浓缩。通过柱色谱(硅胶,10∶1 CH2Cl2/MeOH,8∶2∶0.2 CHCl3/CH3OH/NH4OH)纯化残余物,得到作为黄色固体的化合物XV-cc(1.24g,55%)。
1H NMR(400MHz,CD3OD):δ7.45-7.39(m,4H),7.35-7.24(m,6H),6.88(s,2H),5.47(s,2H),4.21(dd,J=10.7,5.3Hz,2H),4.00-3.89(m,4H),3.85(dd,J=5.1,2.5Hz,2H),3.70(dd,J=9.3,2.5Hz,2H),3.57(t,J=10.7Hz,2H),3.24(t,J=6.8Hz,2H),3.08(t,J=6.4Hz,1H),2.74-2.41(m,16H),1.80-1.70(m,6H),1.71-1.52(m,6H).
3,5-二氨基-N-(N-(4-(4-((S)-4-氨基-3-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)丙基氨基)-4-氧代丁基)-5,6,7,8-四氢萘-1-基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(XV-dd):
向化合物XV-cc(1.24g,1.15mmol)中添加4N HCl水溶液(50mL),并在室温下将混合物搅拌2h。除去溶剂并且通过C18反相Gold柱纯化残留物,得到作为黄色吸湿性固体的化合物XV-dd(700mg,61%)。
1H NMR(400MHz,DMSO-d6):δ10.50(brs,1H),9.67-9.54(m,1H),9.25(t,J=5.4Hz,1H),9.14-9.02(m,1H),8.94-8.87(m,2H),8.86-8.72(m,1H),8.23(brs,1H),7.81(brs,2H),7.42(s,3H),6.91(ABq,J=7.8Hz,2H),5.54-5.38(m,1),5.02-4.22(m,3H),4.11-4.00(m,2H),3.95-3.84(m,1H),3.71(d,J=5.0Hz,2H),3.59(dd,J=10.7,2.4Hz,2H),3.54-3.44(m,5H),3.44(dd,J=10.9,5.4Hz,2H),3.42-3.16(m,13H),3.03-2.82(m,2H),2.70-2.58(m,3H),2.54(d,J=9.4Hz,1H),2.23-2.10(m,2H),2.09-2.06(m,1H),2.00-1.90(m,1H),1.76-1.67(m,4H),1.65-1.48(m,5H),
1H NMR(400MHz,CD3OD):δ6.94(s,2H),4.25-4.18(m,2H),4.03(t,J=6.2Hz,1H),3.86(d,J=4.6Hz,2H),3.78(dd,J=10.6,2.3Hz,2H),3.75-3.62(m,6H),3.61-3.51(m,2H),3.50-3.41(m,4H),3.36(t,J=7.3Hz,2H),3.21-3.09(m,2H),2.77-2.68(m,4H),2.68-2.60(m,4H),2.33-2.21(m,2H),2.16-2.05(m,2H),1.83-1.76(m,4H),1.75-1.71(m,2H),1.70-1.61(m,2H).
HRMS,C39H66ClN10O12[M+H]+计算值:901.4506;实测值:901.4553。
方案XIV.3,5-二氨基-N-(N-(4-((S)-3-氨基-2-(3-(双((2S,3R,4R,5R)-2,3,4,5,6-五羟基己基)氨基)-3-氧代丙基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺(XIV-ee)的合成
方案XVII.3,5-二氨基-N-(N-(4-(4-((S)-3-氨基-2-(3-(己基((2S,3R,4R,5R)-2,3,4,5,6五羟基)氨基)丙基氨基)-3-氧代丙基)苯基)丁基)甲脒基)-6-氯吡嗪-2-甲酰胺的盐酸盐(XVII-d)的合成:
一些测定可用于表征本发明的化合物。代表性的测定将在下面讨论。
测定1.钠通道体阻断活性和可逆性的体外测量
一种用于评估本发明化合物的作用机制和/或效力的测定包括使用在尤斯灌流室(Ussing Chamber)上安装的气道上皮单层在短路电流(ISC)下测量气道上皮钠电流来测定腔内药物抑制。该试验详细描述在Hirsh,A.J.,Zhang,J.,Zamurs,A等Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carbonyl)-N’-4-[4-(2,3-dihydroxyprop oxy)pheny]butyl-guanidine methanesulfonate(552-02),a novel epithelial sodium channel blocker with potential clinical efficacy for CF lung disease.J.Pharmacol.Exp.Ther.2008;325(1):77-88中。将获自新鲜切除的人、狗、绵羊或啮齿动物气道的细胞接种到多孔的0.4微米SnapwellTM Inserts(CoStar)上,在激素限定的培养基中在空气-液体界面(air-liquid interface,ALI)条件下培养,在浸入尤斯灌流室中的Krebs Bicarbonate Ringer(KBR)时测定钠转运活性(ISC)。以半对数(half-log)剂量添加方案向腔内浴(lumenal bath)中添加所有受试化合物(1×10-11M至3×10-5M),并且记录ISC(抑制)的累积变化。在二甲基亚砜中制备所有药物以1×10-2M浓度作为储液并储存在-20℃下。通常平行地进行8份制备;每进行两份制备物引入阿米洛利和/或苯扎米尔作为阳性对照。在施用最大浓度(5×10-5M)之后,用新鲜的无药物KBR溶液置换腔内浴三次,并且在每次清洗约5分钟的持续时间之后测量所得ISC。可逆性定义为在第三次清洗后钠电流返回基线值的百分比。通过计算机接口收集来自电压钳的所有数据并离线分析。
通过Prism 3.0程序考虑和分析所有化合物的剂量效应关系。计算IC50值、最大有效浓度和可逆性并与作为阳性对照的阿米洛利和苯扎米尔进行比较。从犬气道新鲜切除的细胞中的代表性化合物相对于阿米洛利的钠通道阻断活性的效力示于表1中。
表1.在犬支气管上皮(canine bronchial epithelial,CBE)细胞中通过化合物(Ia)抑制短路电流(IC50nM)
测定2.在绵羊中黏液纤毛清除(Mucociliary Clearance,MCC)研究
在MCC中最经常用来进行测量的动物模型是绵羊模型。可使用由Sabater等,Journal of Applied Physiology,1999,第2191至2196页(其通过引用并入本文)描述的体内模型测量化合物对增强黏膜纤毛清除(MCC)的作用。
在这些研究中,控制成年绵羊并经鼻插入气管插管。对绵羊施用10-15分钟的雾化试验样品。然后,在试验样品之后指定时间的四小时或八小时施用放射性标记99mTc-硫胶体(TSC,3.1mg/mL;含约20mCi)。通过气管插管施用放射性标记的气雾剂约5分钟。然后将绵羊的插管拔除,并在1小时的观察期内,每5分钟测量一次肺中的总放射性计数。肺中放射性的清除率代表了动物中的MCC速率。该系统的优点在于,它近似地模拟了人的肺环境。该模型还允许在测试期间通过血浆和尿液取样同时收集PK/PD信息。还有几种技术可在MCC的测量期间测量气道表面的药物浓度。这些技术包括收集呼出气冷凝物或通过支气管镜检由滤纸法获得ASL。
上述的绵羊模型用于评估气雾剂递送的化合物II-d对MCC的体内作用(效力/耐久性)。处理由化合物的任一4毫升的化合物II-d、比较实施例1、(S)-3,5-二氨基-6-氯-N-(N-(4-(4-(2,3-二氨基-3-氧代丙氧基)苯基)丁基)甲脒基)吡嗪-2-甲酰胺、载剂(无菌蒸馏水)中的任一种组成,或测试与HS组合的测试剂。为了确定是否已将HS与化合物II-d MCC组合,要在施用化合物II-d之后立即施用HS。使用雨滴喷雾器(Raindrop nebulizer)以每分钟八升的流速雾化试验溶液,并连接到由电磁阀和压缩空气源(20psi)组成的一个剂量测定系统上。在使用雨滴喷雾器进行气雾剂施用之后,药物在绵羊肺中的沉积剂量估计为剂量的8%至15%。用雨滴喷雾器,在药物处理后的4或8小时,施用放射性标记的TSC约3分钟以评价其效力/耐久性。用γ照相机在右肺的中心区域每间隔5分钟测量一次放射性计数,进行一小时。利用三种分析方法,1)使用线性回归拟合的在前30分钟内清除的初始速率(斜率),2)在一小时的时间内%清除曲线下面积,以及3)在一小时内得到的最大清除率。
测试了在给药后4小时,化合物II-d在0.024nmol/kg,0.24nmol/kg和2.4nmol/kg对绵羊MCC的作用,并与载剂(4mL无菌H2O)进行了比较(图1)。作用的分析示于表2。与载剂对照相比,所有的测试剂量的化合物II-d都增强了MCC。认为0.24nmol/kg的剂量为最大值(100%)的MCC作用,如图2所示其在剂量响应曲线的顶部,化合物II-d的ED50剂量是约0.024nmol/kg。重要的是,如图2所示剂量高达24nmol/kg(ED50的1,000倍ED50)没有表现出提高了血浆中钾(高钾血症的标志)。
表2.在化合物(Ia)或载剂给药后4h时绵羊的MCC
数据表示为平均值+SD(n),*表示与载剂相比的显著性(p<0.05)。
图3表明萘基系列的化合物中也存在对绵羊MCC的强大体内作用。
测定3.通过人气道上皮清除和代谢气道表面药液(ASL)
评估了在人支气管上皮(HBE)细胞中化合物II-d从顶端的消失和气道上皮的代谢(表3)。在这些实验中,将25μL的25μM ENaC阻断剂的溶液添加到在气/液界面生长的HBE细胞的顶端表面,并且通过UPLC测量了2h内顶端和基底侧室中的药物浓度。在化合物II-d在顶端表面孵育2h后(37℃),在顶端或基底侧都没有检测到代谢物,且在基底侧没有检测到化合物II-d。
表3.化合物II-d以及相关化合物与比较实施例1在HBE的顶端消失和代谢
数值表示为平均值±SD
测定4.气道水化和钠通道阻断(体外模型)
Parion Sciences已经了开发了用于评估细胞培养物中气道水化的实验模型(Hirsh,A.J.,Sabater,J.R.,Zamurs,A等,Evaluation of second generation amiloride analogs as therapy for CF lung disease.J.Pharmacol.Exp.Ther.2004;311(3):929-38.Hirsh,A.J.,Zhang,J.,Zamurs,A等,Pharmacological properties of N-(3,5-diamino-6-chloropyrazine-2-carb onyl)-N′-4-[4-(2,3-dihydroxy propoxy)phenyl]butyl-guanidine methane sulfonate(552-02),a novel epithelial sodium channel blocker with potential clinical efficacy for CF lung disease.J.Pharmacol.Exp.Ther.2008;325(1):77-88)。
将原代CBE细胞接种到在空气-液体界面保持的包被胶原的多孔膜上,以评估随着时间的推移表面液体体积的维持。在实验开始时,从含有空气-液体界面培养基的板中移走12mm的卡孔插件(snapwell insert),吸干,称重并将50μL的载剂(0.1%DMSO)或ENaC阻断剂(10μM,于0.1%的DMSO中)施加到顶端表面并记录质量。将插件立即返回到Transwell平板(500μL,Krebs Ringer Bicarbonate(KRB),pH 7.4,在下部腔室中),并放置在37℃,5%CO2的培养箱中。为了降低由于顶部碳水化合物渗透梯度对失水的假象,在顶部缓冲液中不包括葡萄糖。对化合物(1a)进行了测试并与载剂进行了比较,并且连续测量了0至8或24小时内ASL的质量。将表面液体的质量转化为按μL计的体积。数据表示为初始体积%(100%=50μL)。
通过测定将50μL体积的实验缓冲液添加到CBE细胞的顶端表面之后保留的缓冲液,间接地确定钠转运抑制的持续时间。8小时后,仅有12.5±12.1%的载剂(缓冲液)保留在表面上,并且在载剂中加入10μM阿米洛利(25±19.2%,8小时后)表面液体滞留有小幅提高。相比之下,化合物II-d显著提高了顶端表面液体的滞留,在8小时内保持了112±11%的表面液体。
为了进一步测试化合物II-d,将孵育的持续时间从8小时增至24小时。对阿米洛利的测试未超过24小时,因为其主要效果在8小时后消失。24小时后,阿米洛利仅保持了11%的载剂缓冲液,而化合物II-d在24小时内保持了70.6±8.0%(n=42)的表面液体,相对于8小时的测量仅仅损失了16%,这表明化合物II-d表现出持久保持液体的作用。
比较实施例
与已知的钠通道阻滞剂例如阿米洛利和如下文比较实施例1中所述的第三代化合物相比,本发明式(A)的化合物更强大和/或从黏膜表面(特别是气道表面)更不迅速地吸收。因此,如表4所示的数据所证明的,与这些已知的化合物相比,式(A)化合物在黏膜表面有更长的半衰期。评估了在HBE中化合物II-d从顶端表面的消失和气道上皮的代谢,并与对比实施例1进行了比较(表4)。在这些实验中,将25μL的25μM ENaC阻断剂的溶液添加到在气/液界面培养的HBE细胞的顶端表面,并且通过UPLC测量了2h内顶端和基底侧室中的药物浓度。在化合物II-d在顶端表面孵育2h后(37℃),在顶端侧或基底侧都没有检测到代谢物,且在基底外侧上仅检测到少量的化合物II-d。与此相反,大多数比较实施例1从顶侧,83%代谢为较低活性的羧酸(S)-2-氨基-3-(4-(4-(3-(3,5-二氨基-6-氯吡嗪-2-羰基)胍基)丁基)苯氧基)丙酸,结构如下。
表4.在HBE中化合物II-d和相关化合物相对于比较实施例1的顶端消失和代谢
数值表示为平均值±SD
比较实施例1,(S)-3,5-二氨基-6-氯-N-(N-(4-(4-(2,3-二氨基-3-氧代丙氧基)苯基)丁基)甲脒基)吡嗪-2-甲酰胺,具有以下结构:
在WO 2003/070182(美国专利号6,858,615;7,186,833;7,189,719;7,192,960;和7,332,496)中要求保护、描述或公开了该结构作为具有可用药用性的钠通道阻断剂并且可通过在其中描述的方法以及本领域公知的其他方法来制备。
可以在美国2005/0080093的第15页上看到在比较实施例1的化合物并作为WO 2008/031048第90页上的化合物2以及作为WO 2008/031028第42-43页的化合物2。为了在治疗囊性纤维化和C.O.P.D中具有可用的活性,化合物必须在基于多次给药而不提高血浆钾(其最终导致高钾血症,一种严重且危险的病症)的剂量下,具有导致增强黏膜纤毛清除(MCC)的性质。因此,必须避免这类化合物,已知如果它们由肾脏显著排泄,则会提高血浆钾。为了评估这种可能性,具有体内的MCC活性是有益的并且在可用剂量下,不会引起血浆钾的升高。一种评估模型是下面描述的绵羊MCC模型。
如从表5和图4中可以看出,使用三种不同的测量方法(斜率,AUC和最大清除率)得到的在绵羊MCC模型中比较实施例1的ED50为约240nmol/kg(3mM)。在这个剂量下(这将是临床活性剂量),比较实施例1导致血浆钾的升高(图5),这在重复给药时将导致高钾血症。因此,比较实施例I不可供人类使用,而化合物II-d产生安全且有效的MCC且在该模型中效益风险比大于1000。
表5.给药后4小时在绵羊中载剂、比较实施例1或化合物II-d的MCC
图6绘制了如以上在MCC模型中所述的化合物II-d和比较实施例1在一段时间的黏液清除百分比的图。化合物II-d在比所见的比较实施例低10,000倍的剂量时提供了类似的黏液清除百分比。化合物II-d在临床相关剂量范围内提供了最大作用。
图7示出了在MCC的研究中,在接受比较实施例1的绵羊血浆中,在有效剂量时可看出血浆钾水平的显著提高。在绵羊的MCC中,化合物II-d与比较实施例1相比,更有效10000倍,且剂量高达24nmol/kg(ED50剂量的1000倍)时也没有升高血浆K。而比较实施例1在约3mM的ED50剂量时就升高了血浆K(图6和7)。这再次表明化合物II-d具有独特且出人意料的效力和安全性优势,如表6中所示,与比较实施例1相比,具有10,000至100,000倍更高的肾脏安全性。
表6.治疗比(效益/风险)
- 还没有人留言评论。精彩留言会获得点赞!