制备2,3,3,3-四氟丙烯的方法与流程
制备2,3,3,3-四氟丙烯的方法本申请是申请日为2009年5月15日、申请号为200980117398.4、发明名称为“制备2,3,3,3-三氟丙烯的方法”(PCT/GB2009/001263,进入国家阶段日期2010年11月15日)之申请的分案申请。技术领域本发明涉及制备2,3,3,3-四氟丙烯的方法。
背景技术:
2,3,3,3-四氟丙烯也称为HFO-1234yf、HFC-1234yf或简称为1234yf。下文中,除非另行说明,否则2,3,3,3-四氟丙烯称为1234yf。已知的制备1234yf的方法通常受困于诸如低产率、和/或处理有毒和/或昂贵的反应物、和/或使用极端条件、和/或产生有毒副产物的缺点。在例如JournalFluorineChemistry(82),1997,171-174中已经描述了制备1234yf的方法。在这篇论文中,通过四氟化硫与三氟乙酰丙酮反应制备1234yf。但是,由于处理反应物的危险及它们的价格,因此这种方法仅具有学术价值。在US-2931840中描述了另一种制备1234yf的方法。在这种情况下,声称在存在或不存在四氟乙烯的情况下热解C1含氯氟烃以制造1234yf。但是,所描述的产率非常低,而且也必须在极端条件下处理危险的化学品。还预计此类方法会产生多种剧毒的副产物。除了克服已知方法的缺点外,希望的是提供一种仅使用易得原料制备1234yf的新方法。本说明书中对之前公开的文献的列举或论述不应必须视为承认该文献是现有技术的一部分或是公知常识。
技术实现要素:
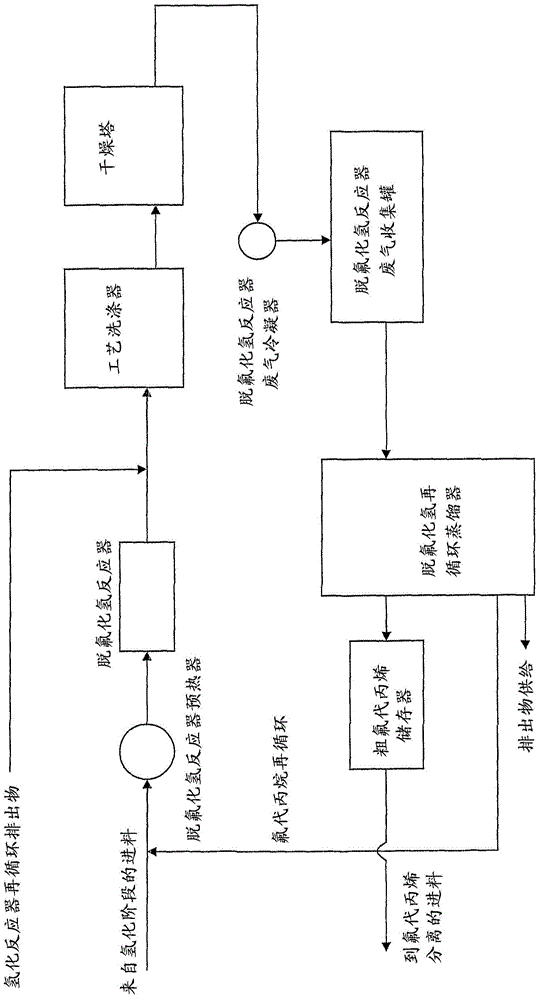
本发明通过提供一种包括以下步骤的制备1234yf的方法解决了已知的制备1234yf方法的缺点:(a)在氢化催化剂的存在下使1,1,2,3,3,3-六氟丙烯(下文中称为1216或HFP)与氢接触,以产生1,1,2,3,3,3-六氟丙烷(下文中称为236ea);(b)使236ea脱氟化氢以产生1,2,3,3,3-五氟丙烯(下文中称为1225ye);(c)在氢化催化剂的存在下使1225ye与氢接触,以产生1,2,3,3,3-五氟丙烷(下文中称为245eb);和(d)使245eb脱氟化氢以产生1234yf。除非另行说明,否则该方法在下文中称为本发明的方法。1225ye以几何异构体E-1225ye和Z-1225ye形式存在。除非另行说明,否则本文中所用的1225ye应当是指几何异构体的混合物。可以使用任何合适的装置,如静态混合器、管式反应器、搅拌釜式反应器或搅拌气液分离容器间歇或连续(优选连续)进行步骤(a)至(d)的每一步骤。装置优选由一种或多种耐腐蚀材料,例如或制造。在本文中所述的本发明方法的任意方面,可以对来自步骤(a)、(b)、(c)和/或(d)的产物进行纯化步骤。例如,可以通过一个或多个蒸馏、浓缩或相分离步骤和/或通过用水或碱水溶液洗涤以分离所需产物或反应物来实现纯化。可以采用多种合适的反应器布局进行本发明的方法。例如,可以使用对于各步骤独立的反应器连续进行该方法,其中步骤(a)、(b)、(c)和(d)以该顺序依次进行。或者,可以使用一个氢化反应器和一个脱氟化氢反应器以半间歇方式进行该方法,其中步骤(a)、(b)、(c)和(d)以该顺序依次进行。在这种半间歇工艺中,在氢化反应器中将HFP转化为236ea,在脱氟化氢反应器中将236ea转化为1225ye。这两个反应,步骤(a)和(b)均进行规定的时间,通常为大约1至大约1000小时,如大约10至大约500小时,例如大约20至大约200小时。在分别使用同一氢化反应器与脱氟化氢反应器以将1225ye转化为245eb和将245eb转化为1234yf之前,将产生的1225ye储存在缓冲罐中。这些反应,步骤(c)和(d)再次进行规定的时间,通常大约1至大约1000小时,如大约10至大约500小时,例如大约20至大约200小时。在另一个优选的反应布局中,本发明方法的步骤(a)和(c)可以在同一反应器中同时进行。氟代烯烃如HFP和1225ye的氢化已知是大量放热的。通过结合氢化反应,认为可以通过利用不同的反应热和产物的热容量来控制反应的放热性质。这对于本发明的方法来说具有较低的资本成本和提高的效率的优点。其中步骤(a)和(c)在同一反应器中同时进行的反应产物包含236ea和245eb。它们可以在送入用于进行步骤(b)和(d)的单独的脱氟化氢反应器中之前彼此分离(例如,通过蒸馏)。或者,在任意任选的去除不需要的副产物(例如CF3CFHCH3(254eb)和/或H2)的纯化步骤之后,可以将236ea和245eb的组合流送入单个反应器中。由此,可以在同一反应器中同时进行脱氟化氢步骤(b)和(d)。据信这对于本发明的方法来说具有较低的资本成本和提高的效率的优点。当然,可以将来自于单独氢化反应器的236ea与245eb的单独进料送入单个脱氟化氢反应器中,在该脱氟化氢反应器中同时进行步骤(b)和(d)。在又一个实施方案中,可以利用稀释气流来控制步骤(a)和/或(c)、特别是步骤(a)的放热氢化反应。为避免疑问,稀释气流可以单独用于步骤(a)、单独用于步骤(c)或用于步骤(a)与(c)。稀释气流可以是气体,如氮气或1,1,1,2-四氟乙烷(134a),过量的一种或多种原料(例如HFP和/或1225ye),或实际上来自步骤(a)和(c)的产物,245eb或236ea的一种或两种。对于步骤(a)和(c)的优选条件、催化剂等的下列描述适用于可用于进行本发明方法的所有反应器布局(例如上述那些)。步骤(a)和(c)中的氢化反应可以在液相或气相,优选气相中进行。大约-50至大约275℃的温度可用于步骤(a)和(c)。液相氢化的优选温度为大约-50至大约50℃,例如大约15至大约40℃。气相氢化的优选温度为大约0至大约250℃,如大约20至大约200℃,例如大约50至大约150℃。步骤(a)和(c)可以在氟化极性非质子溶剂的存在下进行,特别是当在液相中进行时。合适的溶剂包括HFC(例如134a)和PFC(例如全氟萘烷)。步骤(a)和(c)中的氢化反应可以在大气压力、低于大气压力或超大气压力,优选超大气压力下进行。例如,可以在大约0至大约40巴(绝对压力),如大约1至大约30巴(绝对压力),例如大约5至大约20巴(绝对压力)的压力下进行氢化。步骤(a)中氢:1216和步骤(c)中氢:1225ye的比合适地为大约0.1∶1至大约40∶1,如大约1∶1至大约20∶1,优选大约1.1∶1至大约10∶1,例如1.5∶1至大约5∶1。任何合适的氢化催化剂可用于步骤(a)和(c),包括包含过渡金属的催化剂。优选的过渡金属氢化催化剂包括包含Ni、Pd、Pt、Re、Rh、Ru及其混合物的那些催化剂。此类催化剂可以是负载的(例如,负载在氧化铝、二氧化钛、二氧化硅、氧化锆(或前述元素的氟化物)、氟化钙、碳或硫酸钡上)或未负载的(例如雷尼镍或海绵钯)。钯碳(Pd/C)目前是优选的用于步骤(a)和(c)的氢化催化剂。氢化催化剂通常以构成步骤(a)和(c)的成分总重量的大约0.01至大约30重量%,如大约0.1至大约10%的量使用。当Pd/C用作催化剂时,Pd以该催化剂的大约0.01至大约10重量%,如大约0.1至大约5%的量存在。在步骤(a)和(c)中氢和催化剂与1216和1225ye的接触时间合适地为大约1至大约200秒,如大约2至大约150秒。用于步骤(b)和(d)的优选条件、反应物、催化剂等的下列描述适用于可用于进行本发明方法的所有反应器布局(例如上述那些)。本发明方法的步骤(b)和(d)可以在有效地使236ea脱氟化氢以产生1225ye和/或使245eb脱氟化氢以产生1234yf的任何合适的反应条件下进行。脱氟化氢可以在气相或液相中并在大约-70至大约1000℃(例如大约0至大约400℃)的温度下进行。该工艺可以在大气压力、低于大气压力或超大气压力,优选大约0至大约30巴(绝对压力)下进行。脱氟化氢反应可以是热引发的,可以是碱介导的和/或可以是通过任何合适的催化剂催化的。合适的催化剂包括基于金属和碳的催化剂,如包含活性碳、主族(例如基于氧化铝的催化剂)和过渡金属的那些,如基于氧化铬的催化剂(例如锌/氧化铬)或基于镍的催化剂(例如镍网)。在步骤(b)和(d)中实现脱氟化氢的一种优选方法是使236ea和245eb与基于氧化铬的催化剂接触,该催化剂为例如在EP-A-0502605、EP-A-0773061、EP-A-957074、WO98/10862和WO2006/106353(例如,锌/氧化铬催化剂)中描述的那些。术语“锌/氧化铬催化剂”指的是包含铬或铬化合物和锌或锌化合物的任何催化剂。通常,存在于本发明的锌/氧化铬催化剂中的铬或铬化合物是铬的氧化物、氟氧化物或氟化物,如铬的氧化物。存在于本发明的锌/氧化铬催化剂中的锌或锌化合物的总量通常为大约0.01%至大约25%、优选0.1%至大约25%、合适地为0.01%至6%的锌,并且在一些实施方案中,优选为该催化剂的0.5重量%至大约25重量%,优选为该催化剂的大约1至10重量%,更优选为该催化剂的大约2至8重量%,例如该催化剂的大约4至6重量%。在其它实施方案中,该催化剂合适地包含0.01%至1%、更优选0.05%至0.5%的锌。优选的量取决于许多因素,如铬或铬化合物和/或锌或锌化合物的性质和/或制备催化剂的方法。下面更详细地描述这些因素。要理解的是,本文中提到的锌或锌化合物的量指的是元素锌的量,无论以元素锌形式还是以锌化合物的形式存在。本发明中所用的锌/氧化铬催化剂可以包括额外的金属或其化合物。通常,该额外的金属是二价或三价金属,优选选自镍、镁、铝及其混合物。通常,该额外的金属以催化剂的0.01重量%至大约25重量%、优选催化剂的大约0.01至10重量%的量存在。其它实施方案可以包含至少大约0.5重量%或至少大约1重量%的额外金属。本发明中所用的锌/氧化铬催化剂可以是非晶的。这意味着当通过例如X射线衍射分析时催化剂不显示显著的结晶特征。或者,催化剂可以是部分结晶的。这意味着0.1至50重量%的催化剂为一种或多种结晶铬化合物和/或一种或多种结晶锌化合物的形式。如果使用部分结晶催化剂,则其优选含有0.2至25重量%、更优选0.3至10重量%、再更优选0.4至5重量%的一种或多种结晶铬化合物和/或一种或多种结晶锌化合物形式的催化剂。在用于脱氟化氢反应的过程中,结晶度可以改变。因此,可能的是,本发明的催化剂在用于脱氟化氢反应之前具有如上规定的结晶度,而在用于脱氟化氢反应的过程中或之后具有在这些范围之外的结晶度。在本发明的催化剂中结晶材料的百分比可以通过本领域已知的任何合适方法测定。合适的方法包括X射线衍射(XRD)技术。当采用X射线衍射时,结晶材料的量,如结晶铬氧化物的量可以参照存在于催化剂中的已知量的石墨(例如用于制备催化剂丸粒的石墨)测定,或更优选通过比较样品材料与由适当的国际承认的标准物制得的参比材料,例如NIST(NationalInstituteofStandardsandTechnology)参比材料的XRD图谱的强度来测定。本发明的锌/氧化铬催化剂在用含氟化物物质,如氟化氢或氟化烃对其进行预处理之前通常具有至少50米2/克、优选70至250米2/克和最优选100至200米2/克的表面积。在该预处理(下文中进行更详细地描述)过程中,催化剂中的至少部分氧原子被氟原子替代。本发明的锌/氧化铬催化剂通常具有活性与选择性水平的有利平衡。它们优选还具有一定程度的化学鲁棒性,这意味着它们具有相对长的工作寿命。本发明的催化剂优选还具有使得能够相对容易处理的机械强度,例如可以采用已知技术将它们装入到反应器中或从反应器中排出。可以以本领域已知的任何合适的形式提供本发明的锌/氧化铬催化剂。例如,它们可以以适用于固定床或流化床的尺寸的丸粒或颗粒形式提供。催化剂可以是负载的或未负载的。如果催化剂是负载的,则合适的载体包括AIF3、氟化的氧化铝或活性碳。本发明的锌/氧化铬催化剂包括此类催化剂的升级形式(promotedform),包括含有提高的路易斯和/或酸性和/或碱性的那些。可用于本发明的非晶催化剂可以通过任何用于制备基于氧化铬的非晶催化剂的本领域已知方法获得。合适的方法包括在加入氢氧化铵时从锌和铬的硝酸盐溶液中共沉淀。或者,可以采用将锌或其化合物表面浸渍到非晶氧化铬催化剂上。制备非晶锌/氧化铬催化剂的另外的方法包括例如用锌金属将铬(VI)化合物,例如铬酸盐、重铬酸盐、特别是重铬酸铵还原为铬(III),然后共沉淀并洗涤;或以固体形式混合铬(VI)化合物和锌化合物,例如醋酸锌或草酸锌,并将混合物加热至高温以将铬(VI)化合物还原为铬(III)氧化物并将锌化合物氧化为氧化锌。可以以化合物,例如卤化物、卤氧化物、氧化物或氢氧化物(至少一定程度上取决于采用的催化剂制备技术)的形式将锌引入到非晶氧化铬催化剂之中和/或之上。在通过浸渍氧化铬、卤化氧化铬或铬的卤氧化物制备非晶催化剂的情况下,该化合物优选是水溶性盐,例如卤化物、硝酸盐或碳酸盐,并以水溶液或浆料的形式使用。或者,可以将锌和铬的氢氧化物共沉淀(例如通过使用碱,如氢氧化钠或氢氧化铵),随后转化为氧化物以制备非晶催化剂。将不溶性锌化合物与基础氧化铬催化剂混合并研磨提供了制备非晶催化剂前体的另一种方法。制备基于铬卤氧化物的非晶催化剂的方法包括将锌化合物加入到水合铬卤化物中。引入到非晶催化剂前体中的锌或锌化合物的量取决于所采用的制备方法。据信工作催化剂具有位于含铬晶格,例如铬的氧化物、卤氧化物或卤化物晶格中的含锌阳离子表面。因此,所需的锌或锌化合物的量对于通过浸渍制得的催化剂来说通常低于通过其它方法,如共沉淀制得的在非表面位置也含有锌或锌化合物的催化剂。任何前述方法或其它方法可用于制备可用在本发明方法中的非晶催化剂。本文中所述的锌/氧化铬催化剂通常在使用前通过热处理稳定化,使得它们在它们使用时所暴露的环境条件下是稳定的。该稳定化通常是两阶段过程。在第一阶段中,通过在氮气或氮气/空气环境中进行热处理使催化剂稳定。在本领域中,该阶段通常称为“煅烧”。氟化催化剂随后通常通过在氟化氢中进行热处理以对氟化氢稳定。该阶段通常称为“预氟化”。通过小心控制进行这两个热处理阶段的条件,可以使催化剂的结晶度达到受控程度。例如,非晶催化剂可以在合适的气氛中在大约300至大约600℃、优选大约400至600℃、更优选500至590℃,例如520、540、560或580℃的温度下热处理大约1至大约12小时,优选大约2至大约8小时,例如大约4小时。可以在其中进行该热处理的合适气氛包括氮气气氛或氮气中氧含量为大约0.1至大约10v/v%的气氛。或者,也可使用其它氧化性环境。例如,含有合适氧化剂的环境包括但不限于含有硝酸盐、CrO3或O2(例如空气)的源的那些。可以在现有技术中常用的制备非晶催化剂的煅烧阶段之外,或替代该煅烧阶段进行该热处理阶段。可以选择预氟化阶段的条件,使得其引起催化剂结晶度的变化或使得其不会引起这种变化。本发明人已经发现,在氟化氢的存在下,任选在另一种气体如空气的存在下,在大气压力或超大气压力下在大约250至大约500℃、优选大约300至大约400℃的温度下对催化剂前体热处理大约1至大约16小时,可以产生其中结晶度如上定义的催化剂,例如催化剂的0.1至8.0重量%(通常为催化剂的0.1至低于8.0重量%)为一种或多种结晶铬化合物和/或至少一种额外金属的一种或多种结晶化合物的形式。本领域技术人员会认识到,通过改变上述条件,如通过改变进行热处理的温度和/或时间和/或气氛,可以改变催化剂的结晶度。通常,例如可以通过在进行催化剂预处理时升高温度和/或延长煅烧时间和/或增强气氛的氧化性,来制备具有更高结晶度(例如催化剂的8至50重量%)的催化剂。下表描述了催化剂结晶度随煅烧温度、时间和气氛的变化,下表显示了一系列实验(其中在多种条件下对8克6%的锌/氧化铬催化剂进行煅烧)和通过X射线衍射测定的结晶度水平。预氟化处理通常具有降低催化剂表面积的效果。在预氟化处理后,本发明的催化剂通常具有20至200米2/克,如50至150米2/克,例如低于大约100米2/克的表面积。在使用中,可以通过在空气中在大约300℃至大约500℃的温度下加热以周期性再生或再活化锌/氧化铬催化剂。空气可以与惰性气体如氮气,或与氟化氢(热的来自催化剂处理工艺并可直接用于使用再活化催化剂的氟化工艺)混合使用。步骤(b)和(d)中所用的催化剂可以以有机物(例如236ea和/或245eb)重量的大约0.01至大约50重量%,如大约0.1至大约30%,例如大约0.5至大约20%的量使用。236ea和245eb的(金属或碳)催化的脱氟化氢通常在大约0至大约400℃的温度下进行。例如,当在基于氧化铬的催化剂(例如,锌/氧化铬催化剂)的存在下进行时,步骤(b)和(d)优选在大约200至大约360℃,如大约240至大约340℃的温度下进行。步骤(b)和(d)优选在大约0.01至大约25巴(绝对压力)或大约0.1至大约20巴(绝对压力),如大约1至大约10巴(绝对压力)(例如1至5巴(绝对压力))的压力下进行。在步骤(b)和(d)中的催化脱氟化氢中,236ea和/或245eb与催化剂的接触时间合适地为大约1至大约500秒,如大约5至大约400秒。本发明的脱氟化氢步骤(b)和(d)可以在氟化氢(HF)的存在下进行。例如,可以存在由236ea和/或245eb脱氟化氢所形成的HF和/或来自单独进料的HF。在某些实施方案中,希望的是使用一些HF,以防止和/或减缓步骤(b)和(d)中有机进料过度分解和/或催化剂结焦。或者,步骤(b)和(d)可以在不存在HF的情况下进行和/或从反应器中除去HF以帮助促进脱氟化氢反应。当步骤(b)和(d)中存在HF时,HF:有机物(例如,236ea和/或245eb)的摩尔比优选为大约0.01∶1至大约50∶1,如大约0.1∶1至大约40∶1,例如大约0.5∶1至大约30∶1或大约2∶1至大约15∶1(例如大约5∶1至大约10∶1)。在步骤(b)和(d)中进行脱氟化氢的另一种优选方法是使236ea和/或245eb与碱接触(碱介导脱氟化氢)。碱优选是金属氢氧化物或氨基化物(优选碱性金属氢氧化物或氨基化物,例如碱金属或碱土金属氢氧化物或氨基化物)。除非另行说明,否则本文中所用的术语“碱金属氢氧化物”指的是选自氢氧化锂、氢氧化钠、氢氧化钾、氢氧化铷和氢氧化铯的化合物或化合物的混合物。类似地,术语“碱金属氨基化物”指的是选自氨基化锂、氨基化钠、氨基化钾、氨基化铷和氨基化铯的化合物或化合物的混合物。除非另行说明,否则本文中所用的术语“碱土金属氢氧化物”指的是选自氢氧化铍、氢氧化镁、氢氧化钙、氢氧化锶和氢氧化钡的化合物或化合物的混合物。类似地,术语“碱土金属氨基化物”指的是选自氨基化铍、氨基化镁、氨基化钙、氨基化锶和氨基化钡的化合物或化合物的混合物。通常,步骤(b)和(d)的碱介导脱氟化氢工艺在大约-50至大约300℃的温度下进行。优选在大约20至大约250℃,例如大约50至大约200℃的温度下进行该工艺。碱介导脱氟化氢可以在大约0至大约30巴(绝对压力)的压力下进行。碱介导脱氟化氢工艺的反应时间可以在宽范围内改变。但是,反应时间通常为大约0.01至大约50小时,如大约0.1至大约30小时,例如大约1至大约20小时。当然,本领域技术人员会认识到,可以根据多种因素,如所用的碱的性质和/或催化剂的存在等改变进行碱介导脱氟化氢的优选条件(例如温度、压力和反应时间)。步骤(b)和(d)的碱介导脱氟化氢工艺可以在存在或不存在溶剂的情况下进行。如果不使用溶剂,则可以例如在管式反应器中将236ea和/或245eb通入熔融碱或热碱之中或之上。如果使用溶剂,则在某些实施方案中优选的溶剂是水,但是可以使用许多其它溶剂。在某些实施方案中,溶剂如醇(例如丙烷-1-醇)、二醇(例如乙二醇)和多元醇如聚乙二醇(例如PEG200或PEG300)可以是优选的。这些溶剂可以单独使用或联合使用。在其它实施方案中,来自称作极性非质子溶剂类溶剂可以是优选的。此类极性非质子溶剂的实例包括二甘醇二甲醚、环丁砜、二甲基甲酰胺(DMF)、二氧杂环己烷、乙腈、六甲基磷酰胺(HMPA)、二甲亚砜(DMSO)和N-甲基吡咯烷酮(NMP)。溶剂的沸点优选使得其在反应条件下不会产生额外的压力。优选的碱是选自氢氧化锂、氢氧化钠和氢氧化钾的碱金属氢氧化物,更优选氢氧化钠和氢氧化钾,最优选氢氧化钾。另一种优选的碱是选自氢氧化镁和氢氧化钙的碱土金属氢氧化物,更优选氢氧化钙。碱通常以构成步骤(b)和(d)的组分总重量的1至50重量%的量存在。碱优选以5至30重量%的量存在。如上所述,碱介导脱氟化氢可以优选使用水作为溶剂。因此,脱氟化氢反应可以优选使用至少一种碱,如碱(或碱土)金属氢氧化物的水溶液,而不需要助溶剂或稀释剂。但是,可以使用助溶剂或稀释剂来例如改变体系粘度、用作反应副产物的优选相、或提高热质量。可用的助溶剂或稀释剂包括对工艺的平衡或动力学为非反应性的或不会不利地影响工艺的平衡或动力学的那些,包括醇,如甲醇和乙醇;二醇,如乙二醇;醚,如二乙醚、二丁醚;酯,如乙酸甲酯、乙酸乙酯等;直链、支链和环状烷烃,如环己烷、甲基环己烷;氟化稀释剂,如六氟异丙醇、全氟四氢呋喃和全氟萘烷。步骤(b)和(d)的碱介导脱氟化氢优选在催化剂的存在下进行。催化剂优选为相转移催化剂,其促进离子化合物从例如水相转移到有机相中。如果水用作溶剂,则由于碱金属氢氧化物而存在水相或无机相,由于碳氟化合物而存在有机相。相转移催化剂促进这些不同组分的反应。虽然各种相转移催化剂可以以不同方式起作用,但是它们的作用机理并非它们在本发明中效用的决定因素,只要它们促进脱氟化氢反应即可。相转移催化剂可以是离子的或中性的,通常选自冠醚、盐、穴醚和多亚烷基二醇及其衍生物(例如其氟化衍生物)。应使用有效量的相转移催化剂以实现所需反应、影响对所需产物的选择性或提高产率;一旦选择了反应物、工艺条件和相转移催化剂,则可以通过有限次实验确定该量。通常,相对于步骤(b)和(d)中有机化合物的量催化剂的用量为0.001至20摩尔%,如0.01至10摩尔%,例如0.05至5摩尔%。冠醚是其中醚基团通过二亚甲基键连接的环状分子。冠醚形成为据信能够接收或容纳氢氧化物的碱金属离子并由此促进反应的分子结构。特别有用的冠醚包括18-冠-6(尤其与氢氧化钾联用)、15-冠-5(尤其与氢氧化钠联用)和12-冠-4(尤其与氢氧化锂联用)。上述冠醚的衍生物也是可用的,如二苄基-18-冠-6、二环己烷基-18-冠-6、二苄基-24-冠-8和二苄基-12-冠-4。类似于冠醚并可用于相同目的的其它化合物是通过用其它类型的供体原子,特别是N或S替代一个或多个氧原子而不同的化合物。也可使用所有上述化合物的氟化衍生物。穴醚是另一类可在碱介导脱氟化氢中用作相转移催化剂的化合物。穴醚是通过用含有适当隔开的供体原子的链连接桥头结构而形成的三维多大环螯合剂。桥的供体原子可以都是O、N或S,或者化合物可以是混合的供体大环,其中桥缆含此类供体原子组合。合适的穴醚包括用(-OCH2CH2-)基团的链连接氮桥头所得的双环分子,例如[2.2.2]穴醚(4,7,13,16,21,24-六氧杂-1,10-二氮杂双环[8.8.8]二十六碳烷,可以以商品名Kryptand222和Kryptofix222获得)。可以在步骤(iii)的碱介导工艺中用作催化剂的盐包括季盐和季铵盐,其可以分别由式R1R2R3R4p+Z-和R1R2R3R4N+Z-表示。在这些式中,R1、R2、R3和R4各自通常独立地代表C1-10烷基、芳基(例如苯基、萘基或吡啶基)或芳烷基(例如苄基或C1-10烷基取代的苯基),Z-是卤离子或其它合适的反离子(例如硫酸氢根)。此类盐和季铵盐的具体实例包括四甲基氯化铵、四甲基溴化铵、苄基三乙基氯化铵、甲基三辛基氯化铵(可以以商品名Aliquat336和Adogen464购得)、四正丁基氯化铵、四正丁基溴化铵、四正丁基硫酸氢铵、四正丁基氯化四苯基溴化四苯基氯化三苯基甲基溴化和三苯基甲基氯化对于在强碱性条件下使用而言,苄基三乙基氯化铵是优选的。其它可用的盐包括表现出高温稳定性(例如高至大约200℃)的那些盐,例如4-二烷基氨基吡啶盐、四苯基氯化砷双[三(二甲基氨基)膦]氯化亚胺和四[三(二甲基氨基)膦亚胺基]氯化后两种化合物也被报道在热的、浓氢氧化钠存在下是稳定的,因此是特别有用的。可用作相转移催化剂的多亚烷基二醇化合物可以由式R6O(R5O)mR7表示,其中R5是C1-10亚烷基,R6和R7各自独立地为H、C1-10烷基、芳基(例如苯基、萘基或吡啶基)或芳烷基(例如苄基或C1-10烷基取代的苯基),m是至少为2的整数。优选R6和R7是相同的,例如它们都可以是H。此类多亚烷基二醇包括二乙二醇、三乙二醇、四乙二醇、五乙二醇、六乙二醇、二异丙二醇、二丙二醇、三丙二醇、四丙二醇和四亚甲基二醇,单烷基二醇醚如此类二醇的单甲基、单乙基、单丙基和单丁基醚,二烷基醚如四乙二醇二甲醚和五乙二醇二甲醚、此类二醇的苯基醚、苄基醚,以及聚亚烷基二醇如聚乙二醇(平均分子量大约300)和聚乙二醇(平均分子量大约400)和此类聚亚烷基二醇的二烷基(例如二甲基、二丙基、二丁基)醚。也可使用来自上述组之一的相转移催化剂的组合以及来自超过一组的组合或混合物。冠醚和季铵盐是目前优选的催化剂组,例如18-冠-6及其氟化衍生物和苄基三乙基氯化铵。当单独地依次进行步骤(a)至(d)时。每个步骤的产物都可以在不纯化的情况下直接在后继步骤中反应。例如,可以将含有236ea的步骤(a)的产物直接进料到用于脱氟化氢步骤(b)的单独反应器中。步骤(b)甚至可以在与步骤(a)同一反应器中进行,特别是如果对于步骤(a)和(b)使用相同的催化剂更是如此。但是,优选在每个步骤的产物在后继步骤中反应之前将其纯化。可以通过一个或多个蒸馏、浓缩或相分离步骤和/或通过用水或碱水溶液洗涤将每一步骤中所需产物与任何其它产物或反应物分离,由此实现纯化。例如,在进料到脱氟化氢步骤(b)的反应器中之前,可以将步骤(a)中产生的236ea从氢和任何残留的1216中蒸馏出。当步骤(a)和(c)在同一反应器中进行时,可以在不纯化来自步骤(a)和(c)的产物的情况下将来自该反应器的产物进料到一个或多个反应器中以实现步骤(b)和(d)。但是,优选将制得的236ea和245eb与步骤(a)和(c)中的氢和任何残留的1216以及产生的1225ye分离(例如通过蒸馏或任何其它合适的方法),并将其进料到脱氟化氢步骤(b)和(d)的反应器中。如果步骤(b)和(d)不结合的话,则可以在通过单独进行步骤(b)和(d)的反应器之前将236ea和245eb进一步彼此分离。附图说明图1示出了本发明方法的一个优选实施方案的氢化阶段。图2示出了本发明方法的一个优选实施方案的脱氟化氢阶段。图3示出了在本发明方法的一个优选实施方案中1225ye和1234yf的纯化。具体实施方式图1至3示出了本发明方法的一个优选实施方案,其中在同一设备中连续进行氢化步骤(a)和(c)(参见图1),并在同一设备中连续进行脱氟化氢步骤(b)和(d)(参见图2)。如图1中所示,HFP和R1225ye可以与氢一起共进料到氢化反应器中,通常与用于减小由氢化反应导致的放热影响的再循环物一起。也可使用冷却剂来减小放热影响。可以利用通过任何合适装置(例如,鼓风机、压缩机或喷射器)再循环到氢化反应器入口的蒸气将反应器废气部分冷凝。随后可以任选经由氟代丙烷储存/泵送罐将冷凝的液体泵送到蒸馏塔(在图1中称为氢化阶段产物蒸馏器)。含有挥发性较高的氟代丙烯的塔顶馏分通常再循环到氢化反应器中。任选可以调节氢化阶段产物蒸馏器的操作以再循环用于稀释氢化反应器放热的一部分氟代丙烷。还可以取氢化反应器再循环排出物并与脱氟化氢反应器出口结合(参见图2)。可以从塔中以塔底馏分形式取出氢化反应的挥发性较低的氟代丙烷产物(236ea和245eb),(i)以液体形式取出并再蒸发,或(ii)以蒸气形式从蒸馏器底部引出。如图2中所示,蒸发的氢化产物(236ea和245eb)可以与来自脱氟化氢阶段的再循环物混合。在加热至反应温度后,将它们进料到脱氟化氢反应器中。如果需要的话,可以用两个或更多个反应器或反应区域替代该流程图中所示的单个反应器,使得能够优化不同脱氟化氢反应的条件。如上所述,来自脱氟化氢反应器的出口进料可以在去除HF之前与氢化反应器再循环排出物混合,该HF在脱氟化氢反应器中产生并且对于脱氟化氢反应器可以以共同进料形式存在。如图2中所示通过工艺洗涤器,例如通过水洗除去HF。但是,也可以采用其它合适的方法,如共沸蒸馏除去HF。在除去HF并干燥(例如,在干燥塔中用H2SO4)后,随后通常通过废气冷凝器和收集罐将粗产物送至脱氟化氢再循环蒸馏器中。在这里,从该蒸馏器底部取出挥发性较低的未转化的氟代丙烷并将其再循环到反应器中,以塔顶馏分形式从该蒸馏器中取出所需氟代丙烯粗产物(含有1225ye和1234yf)。如图3中所示,通常任选通过氟代丙烯储存/泵送罐将粗氟代丙烯泵送到第一蒸馏塔(图3中标记为1225ye蒸馏器),其以塔底馏分形式除去1225ye用于再循环回到氢化反应中。从蒸馏器中取出含1234yf的塔顶馏分,随后可以将其送入轻物质蒸馏器中,以例如通过除去任何挥发性成分来纯化1234yf。现在通过下列非限制性实施例进一步说明本发明。实施例1:HFP和Z-1225ye的氢化将10克湿的0.5%Pd/C催化剂装入直径大约1.25厘米(0.5”)长20厘米的管式反应器中。将该反应器放在风扇辅助的烘箱中。将热电偶放置在反应器入口和出口处以与催化剂紧密接触。一旦在烘箱中将反应器连到氮气上,将送入氢气和有机物。通过质量流量控制器设置和控制这些进料流。在使用前,首先在110℃下在氮气流(95毫升/分钟)中干燥该催化剂。当两个内部热电偶读数均为大约110℃时,认为催化剂是干的。然后,将氢气(5毫升/分钟)加入到氮气流中并在110℃的温度下保持2小时,由此还原该催化剂。随后将温度升高到150℃再保持30分钟。制备由48.5摩尔%六氟丙烯(1216)和51.5摩尔%Z-3,3,3,2,1-五氟丙烯(Z-1225ye)组成的有机进料混合物。随后使该进料与氢气和氮气的混合物通过该反应器并与该催化剂接触。定期取离开反应器的气体样品,并用GC和GC-MS分析。采用已知标准物校准这些仪器。一系列具有不同进料组成的试验的结果显示在表1中。表1*CF3CFHCH3实施例2:236ea的脱氟化氢将由在氧化铬上6重量%Zn组成的非晶催化剂的2克样品装入到安装在管式炉内的15厘米×1.25毫米反应管中。在250℃下将该催化剂干燥1小时,随后在6∶1的N2:HF比例下在250℃下预氟化1小时,然后将温度升高至380℃,在该温度下停止氮气稀释剂流。大约18小时后,切断HF进料,将反应器冷却至220-240℃。在预氟化后,根据温度与HF:236比例研究236ea的脱氟化氢工艺。选择进料气体流量,使得催化剂与进料混合物之间的接触时间为大约5秒。在0-10的范围内研究HF:236比例。在每一温度下,均使体系平衡大约20分钟,然后在每一温度下取反应器废气样品,以如上文实施例1所述通过GC或GC-MS进行分析。结果显示在表2中。表2实施例3:245eb的脱氟化氢将6克5.2%Zn/氧化铬催化剂装入反应管(1.25厘米(0.5”)×30厘米)中。使用前对该催化剂进行如下预处理。·在3巴(表压)下,在80毫升/分钟氮气下,在250℃下干燥过夜·加热至300℃并在3巴(表压)下用4毫升/分钟HF和80毫升/分钟氮气处理16小时·在300℃下再继续加热4小时的同时,将氮气流减少至0,并保持HF流·保持HF流,以25℃/小时将温度升高至380℃·HF流和380℃下加热再保持3小时预氟化结束时,将反应器温度降至310℃,压力降至5巴(表压),并将HF(大约1-60毫升/分钟)与245eb(大约30-80毫升/分钟)的混合物进料到反应器中。定期取反应器废气样品以通过GC和GC-MS分析。使用已知标准物校准这些仪器。结果显示在下表中。**CF3CF2CH3通过所附权利要求限定本发明。以下内容对应于母案申请中的原始权利要求书,现作为说明书的一部分并入此处:1.一种制备2,3,3,3-四氟丙烯(1234yf)的方法,所述方法包括:(a)在氢化催化剂的存在下使1,1,2,3,3,3-六氟丙烯(1216)与氢接触以产生1,1,2,3,3,3-六氟丙烷(236ea);(b)使236ea脱氟化氢以产生1,2,3,3,3-五氟丙烯(1225ye);(c)在氢化催化剂的存在下使1225ye与氢接触以产生1,2,3,3,3-五氟丙烷(245eb);和(d)使245eb脱氟化氢以产生1234yf。2.项1的方法,其中步骤(a)和(c)在同一反应器中进行。3.项2的方法,其中步骤(a)和(c)同时进行。4.前述项中任一项的方法,其中步骤(b)和(d)在同一反应器中进行。5.项4的方法,其中步骤(b)和(d)同时进行。6.前述项中任一项的方法,其中步骤(a)和(c)在大约-25至大约275℃的温度和大约0至大约40巴(绝对压力)的压力下进行。7.前述项中任一项的方法,其中步骤(a)和(c)在气相中在大约0至大约250℃、优选大约20至大约200℃、更优选大约50至大约150℃的温度下进行。8.前述项中任一项的方法,其中步骤(a)中氢:1216和步骤(c)中氢:1225ye的比例为大约1∶1至大约40∶1。9.前述项中任一项的方法,其中步骤(a)中的氢化催化剂和步骤(c)中的氢化催化剂包含负载或未负载的选自Ni、Pd、Pt、Re、Rh、Ru及其混合物的过渡金属。10.项9的方法,其中所述或每种氢化催化剂负载在氧化铝、二氧化钛、二氧化硅、氧化锆(或前述元素的氟化物)、氟化钙、碳和/或硫酸钡上。11.项9或10的方法,其中所述或每种氢化催化剂是钯碳(Pd/C)。12.前述项中任一项的方法,其中步骤(b)和(d)在-70至1000℃的温度和0至大约30巴(绝对压力)的压力下进行。13.前述项中任一项的方法,其中步骤(b)和(d)在包含活性碳、主族金属和/或过渡金属的催化剂的存在下进行。14.项13的方法,其中步骤(b)和(d)在包含过渡金属的催化剂的存在下进行。15.项14的方法,其中所述包含过渡金属的催化剂包含氧化铬。16.前述项中任一项的方法,其中步骤(b)和(d)在大约0至大约400℃的温度和0.01至大约25巴(绝对压力)的压力,优选在大约200至大约360℃和大约1至大约10巴(绝对压力)下进行。17.项1至12中任一项的方法,其中步骤(b)和(d)在碱的存在下进行。18.项17的方法,其中步骤(b)和(d)在大约-50至大约300℃、优选大约20至大约250℃的温度下进行。19.项17或18的方法,其中所述碱选自金属氢氧化物、金属氨基化物及其混合物。20.项17至19中任一项的方法,其中所述碱是碱金属氢氧化物,所述碱金属氢氧化物优选选自氢氧化钠和氢氧化钾。21.项17至20中任一项的方法,其中所述碱是碱土金属氢氧化物,所述碱土金属氢氧化物优选为氢氧化钙。22.项17至21中任一项的方法,其中步骤(b)和(d)在溶剂中进行,所述溶剂优选选自水、醇、二醇、多元醇、极性非质子溶剂及其混合物,并且所述方法任选在助溶剂或稀释剂的存在下进行。23.项17至22中任一项的方法,其中步骤(b)和(d)在催化剂的存在下进行,优选其中所述催化剂是冠醚或季铵盐。24.一般如本文中所述的任何新方法。25.一般如本文中参照实施例所述的任何新方法。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 生产2,3,3,3‑四氟丙烯的方法以及提纯2‑氯‑1,1,1,2‑四氟丙烷的方法与流程
- 生产2,3,3,3‑四氟丙烯的方法以及提纯2‑氯‑1,1,1,2‑四氟丙烷的方法与流程
- 氯丙烯的制造方法和2,3,3,3-四氟丙烯的制造方法与流程
- 含有2‑氯‑1,3,3,3‑四氟丙烯以及1‑氯‑3,3,3‑三氟丙烯的类共沸组合物的制作方法与工艺
- 一种含有戊吡虫胍和氟烯线砜的杀线虫组合物的制作方法与工艺
- 制备2,3,3,3‑四氟丙烯的方法与流程
- 一种合成2,3,3,3‑四氟丙烯的催化剂及其制备方法和用途与流程
- 一种合成 2,3,3,3‑四氟丙烯的催化剂及其制备方法和用途与流程
- 制备2,3,3,3-四氟丙烯的方法与流程
- 2,3,3,3‑四氟丙烯的纯化方法与流程