用超临界流体色谱制备具有抗病毒活性的新型烟草倍半萜‑H的制作方法与工艺
用超临界流体色谱制备具有抗病毒活性的新型烟草倍半萜-H技术领域本发明属于烟草化学技术领域,具体涉及一种从烟草中首次提取得到的倍半萜类化合物。同时,本发明还涉及该化合物的制备方法和在抗轮状病毒中的应用。
背景技术:
烟草是世界上化学成分最为复杂的植物,次生代谢产物非常丰富,经过几十年的研究,人们目前从烟草中鉴定出来的单体化学物质就超过3000多种,而且还有许多成分尚未鉴定出来。超临界流体色谱是以超临界流体(例如CO2)作为流动相的色谱方法,超临界流体兼有气体的低粘度和高扩散系数,约为液体的10~100倍,又有与液体相近的高密度和强溶解能力,因此超临界流体色谱兼备气相色谱和高效液相色谱两者的优点。它既可以分离液相色谱不易分离的高相对分子质量化合物,也可以分离气相色谱不易分离的的热不稳定、强极性或非挥发性化合物。因此,超临界流体色谱非常适合用于天然产物中化合物的分离制备。倍半萜(sesquiterpenes)是指分子中含15个碳原子的天然萜类化合物。倍半萜类化合物分布较广,在植物体内常以醇、酮、内酯等等形式存在于挥发油中,是挥发油中高沸点部分的主要组成部分。多具有较强的香气和生物活性,是医药、食品、化妆品工业的重要原料。为了研究这类化合物的构效关系,可进一步研究和开发更多的倍半萜类化合物,并从中寻找有效的先导化合物和活性基团。本发明利用超临界流体色谱,从云南烤烟烟叶中分离得到了一种新的倍半萜类化合物,该化合物至今尚未见到相关报道,值得一提的是该化合物具有显著的抗轮状病病毒活性。
技术实现要素:
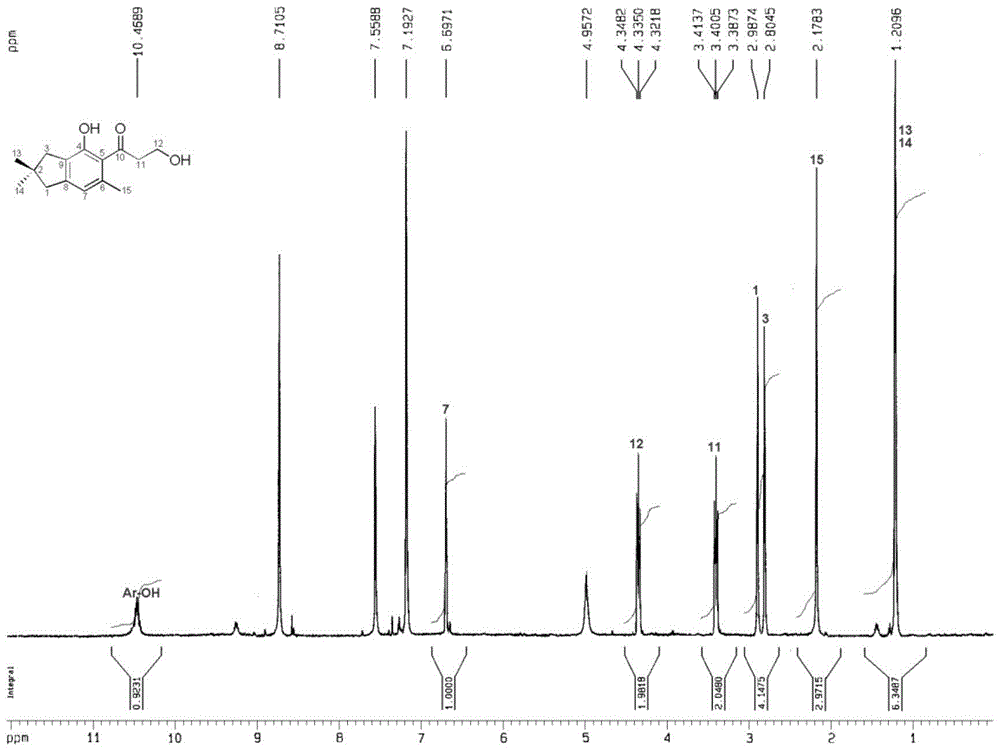
本发明的第一方面在于提供一种新的倍半萜类化合物,该化合物从云南烤烟烟叶中分离得到,其分子式为C15H20O3,经过分析化学鉴定,其具有下述结构:该化合物为浅黄色胶状物,本发明人将其中文名命名为烟草倍半萜-H,其英文名为nicotianasesterpeneH。本发明的第二方面提供了上述第一方面所述的倍半萜类化合物的制备方法,该方法包括如下步骤:(1)制备烟草提取物浸膏:以烟草叶片为原料,将其粉碎或切成小段,并用第一溶剂浸泡并提取所述烟草3~5次,每次24h~72h,将提取液合并、过滤并浓缩后得到所述烟草提取物浸膏;其中所述第一溶剂是选自甲醇、乙醇或丙酮的有机溶剂与水的混合物,其中有机溶剂占该第一溶剂的60wt%~100wt%,且第一溶剂:烟草=2~4:1,重量比;(2)硅胶柱层析:将上述烟草提取物浸膏用选自纯甲醇、纯乙醇或纯丙酮的第二溶剂溶解后与为烟草提取物浸膏的0.8~1.2重量倍的60~120目硅胶拌样,将拌样后的混合物再与为烟草提取物浸膏的2~4重量倍的160~300目硅胶混合后干法装柱,然后用体积比依次为1:0、20:1、9:1、8:2、7:3、6:4、1:1和1:2的氯仿-丙酮溶液进行梯度洗脱,将其中体积为8:2的氯仿-丙酮溶液洗脱时得到的洗脱液称为第一洗脱液。(3)超临界流体色谱分离纯化:将上述第一洗脱液通入超临界流体色谱进行分离纯化,该超临界流体色谱采用10mm×150mm,5μm的Silica2-EP色谱柱,流动相二氧化碳/甲醇(88/12%,质量比),流动相流速为25mL/min,紫外检测器检测波长为254nm,第一洗脱液液每次进样200~500μL,收集每次进样后色谱峰保留时间为15.4min时所对应的洗脱液,称为第二洗脱液,将该第二洗脱液脱除溶剂后即得所述倍半萜化合物。在优选的实施方案中,本发明还包括以下进一步提纯的步骤:将在所述超临界流体色谱分离之后得到的所述倍半萜化合物再次溶于甲醇溶液,并以甲醇溶液的为流动相,通过凝胶柱进行层析分离,提到进一步提纯的所述倍半萜化合物。本发明的第三方面提供了第一方面所述的倍半萜类化合物在制备抗轮状病毒药物中的用途。附图说明图1为本发明倍半萜类化合物的核磁共振碳谱;图2为本发明倍半萜类化合物的核磁共振氢谱;图3为本发明倍半萜类化合物的主要HMBC相关图示。具体实施方式本发明所制备的倍半萜类化合物的结构通过以下方法测定出来的。紫外光谱(溶剂为甲醇),λmax(logε)210(4.22)和257(3.63);红外光谱(溴化钾压片)νmax3386,2928,1657,1600,1546,1433,1158,1064,和936cm-1;高分辨质谱(HRESIMS)给出准分子离子峰m/z247.1327[M-H]-(计算值247.1334)。结合1H和13CNMR谱给出一个分子式C15H20O3,不饱和度为6。其红外光谱显示有羟基(3386cm-1),羰基(1657cm-1)和苯环(1600,1546,1433cm-1)功能团;紫外光谱在257nm处有强吸收,也证实化合物中存在共轭结构。从1H和13CNMR谱(数据归属见表-1)信号可以看出化合物中有一个1,2,3,4,5-五取代的苯环、一个3-羟基-1-丙酮基、三个甲基、两个亚甲基、一个季碳;除去苯环的不饱和度4和羰基的不饱和度1,化合物中还应有一个环。根据H-3和C-2、C-4、C-8、C-9、C-13,14,以及H-1和C-2、C-3、C-7、C-8、、C-13,14的HMBC相关(图-3)可证实C-1、C-2、C-3和C-13,14苯环间形成了一个偕二甲基的5元碳环,该化合物应为偕二甲基二氢茚结构。化合物的母核确定后,剩余的甲基、3-羟基-1-丙酮基和羟基为母核上的取代基。根据H-11和C-5的HMBC相关可证实3-羟基-1-丙酮基取代在母核的C-5位;根据H-15和C-5、C-6和C-7,以及H-7和C-15的HMBC相关,可证实该甲基取代在母核的C-6位;根据酚羟基氢和C-4、C-5和C-9的HMBC相关,可证实酚羟基取代在母核的C-7位。至此本化合物的结构得以确定。表1.化合物的1HNMR和13CNMR数据(C5D5N)本发明化合物是首次被分离出来的,通过上述核磁共振和质谱测定方法确定了为倍半萜类化合物,并表征了其具体结构。经对抗轮状病毒的实验,其TC50值为216.8μg/mL、IC50值为6.52μg/mL,治疗指数TI为33.7;其治疗指数超过对照病毒唑的治疗指数19.16;化合物具有很好的抗轮状病毒活性。以上结果揭示了本发明的化合物在制备抗轮状病毒药物中有良好的应用前景。本发明化合物结构简单活性较好,可作为抗轮状病毒药物研发的先导性化合物用于抗轮状病毒药物制剂研发。下面结合实施例及附图对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或改进,均落入本发明的保护范围。本发明所用原料不受地区和品种限制,任何来源地的烟草均可以实现本发明,下面以来源于云南中烟工业有限责任公司的烟草原料对本发明做进一步说明。除非另有说明,本发明中所采用的百分数均为质量百分数。实施例1烟草样品来源于云南玉溪,品种为玉溪K326。将烟草取样2.0kg粉碎以95%的甲醇提取5次,每次提取24h,提取液合并,过滤,减压浓缩成浸膏,得浸膏105g。浸膏用重量比2.0倍量的纯甲醇溶解后用120g的100目粗硅胶拌样,0.4kg的160目硅胶装柱进行硅胶柱层析,用体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1、1:2的氯仿-丙酮梯度洗脱,薄层色谱(TLC)监测合并相同的部分,得到8个部分,其中体积配比为8:2的氯仿-丙酮洗脱部分用沃特斯SFC80Q超临界流体制备色谱分离,以二氧化碳/甲醇为流动相(88/12%,质量比),Silica2-EP(10mm×150mm,5μm)制备柱为固定相,流动相流速为25mL/min,紫外检测器检测波长为254nm,每次进样200μL,收集每次进样后色谱峰保留时间为15.4min时所对应的洗脱液,多次累加后蒸干,即得本发明所述的倍半萜化合物粗品;该粗品再用纯甲醇溶解,以纯甲醇为流动相,用葡聚糖凝胶柱层析纯化可得纯品。实施例2烟草样品来源于云南大理,品种为云烟200,将烟草取样3.5kg切碎,以95%的乙醇提取4次,每次提取48h,提取液合并,过滤,减压浓缩成浸膏,得浸膏250g。浸膏用重量比2.0倍量的纯甲醇溶解后用250g的80目粗硅胶拌样,0.5kg的200目硅胶装柱进行硅胶柱层析,用体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1、1:2的氯仿-丙酮梯度洗脱,TLC监测合并相同的部分,得到8个部分,其中体积配比为8:2的氯仿-丙酮洗脱部分用沃特斯SFC80Q超临界流体制备色谱分离,以二氧化碳/甲醇为流动相(88/12%,质量比),Silica2-EP(10mm×150mm,5μm)制备柱为固定相,流动相流速为25mL/min,紫外检测器检测波长为254nm,每次进样200μL,收集每次进样后色谱峰保留时间为15.4min时所对应的洗脱液,多次累加后蒸干,即得本发明所述的倍半萜化合物粗品;该粗品再用纯甲醇溶解,以纯甲醇为流动相,用SephadexLH-20凝胶柱层析纯化可得更高纯度的该新化合物。实施例3烟草样品来源于云南昆明,品种为红花大金元,将烟草取样5kg粉碎,以75%的丙酮用超声提取3次,每次提取72h,提取液合并,过滤,减压浓缩成浸膏,得浸膏380g。浸膏用重量比1.6倍量的纯甲醇溶解后用400g的90目粗硅胶拌样,2.4kg的180目硅胶装柱进行硅胶柱层析,用体积配比为1:0、20:1、9:1、8:2、7:3、6:4、1:1、1:2的氯仿-丙酮梯度洗脱,TLC监测合并相同的部分,得到8个部分,其中体积配比为8:2的氯仿-丙酮洗脱部分用沃特斯SFC80Q超临界流体制备色谱分离,以二氧化碳/甲醇为流动相(88/12%,质量比),Silica2-EP(10mm×150mm,5μm)制备柱为固定相,流动相流速为25mL/min,紫外检测器检测波长为254nm,每次进样200μL,收集每次进样后色谱峰保留时间为15.4min时所对应的洗脱液,多次累加后蒸干,即得本发明所述的倍半萜化合物粗品;该粗品再用纯甲醇溶解,以纯甲醇为流动相,用SephadexLH-20凝胶柱层析纯化可得更高纯度的该新化合物。实施例4取实施例1制备的化合物,为浅黄色胶状物。本发明所制备的倍半萜类化合物的结构通过以下方法测定出来的。紫外光谱(溶剂为甲醇),λmax(logε)210(4.22)和257(3.63);红外光谱(溴化钾压片)νmax3386,2928,1657,1600,1546,1433,1158,1064,和936cm-1;高分辨质谱(HRESIMS)给出准分子离子峰m/z247.1327[M-H]-(计算值247.1334)。结合1H和13CNMR谱给出一个分子式C15H20O3,不饱和度为6。其红外光谱显示有羟基(3386cm-1),羰基(1657cm-1)和苯环(1600,1546,1433cm-1)功能团;紫外光谱在257nm处有强吸收,也证实化合物中存在共轭结构。从1H和13CNMR谱(数据归属见表-1)信号可以看出化合物中有一个1,2,3,4,5-五取代的苯环、一个3-羟基-1-丙酮基、三个甲基、两个亚甲基、一个季碳;除去苯环的不饱和度4和羰基的不饱和度1,化合物中还应有一个环。根据H-3和C-2、C-4、C-8、C-9、C-13,14,以及H-1和C-2、C-3、C-7、C-8、、C-13,14的HMBC相关(图-3)可证实C-1、C-2、C-3和C-13,14苯环间形成了一个偕二甲基的5元碳环,该化合物应为偕二甲基二氢茚结构。化合物的母核确定后,剩余的甲基、3-羟基-1-丙酮基和羟基为母核上的取代基。根据H-11和C-5的HMBC相关可证实3-羟基-1-丙酮基取代在母核的C-5位;根据H-15和C-5、C-6和C-7,以及H-7和C-15的HMBC相关,可证实该甲基取代在母核的C-6位;根据酚羟基氢和C-4、C-5和C-9的HMBC相关,可证实酚羟基取代在母核的C-7位。至此本化合物的结构得以确定。实施例5取实施例2制备的化合物,为黄色胶状物。测定方法与实施例4相同,确认实施例2制备的化合物为所述的倍半萜类化合物——烟草倍半萜-H。实施例6取实施例3制备的化合物,为黄色胶状物。测定方法与实施例4相同,确认实施例3制备的化合物为所述的烟草倍半萜-H。实施例7取实施例1-3制备的任一倍半萜类化合物进行抗轮状病毒活性试验,试验情况如下:抗轮状病毒采用体外细胞测试法,即样品与病毒同时作用于MA104细胞后,通过Alarmablue法检测样品对病毒感染致细胞死亡的保护作用,从而测定样品对HRV的活性作用。(a)药物的细胞毒性检测MA104细胞在96孔细胞培养板中培养形成单层后,加入不同浓度的样品液,继续培养3天后,更换含Alamarblue的培养液,继续培养2~3小时后检测其530/590nm处的荧光值,从而检测样品对MA104细胞的毒性,并计算半数细胞毒浓度(TC50)。(b)药物抗轮状病毒作用检测MA104细胞在96孔细胞培养板中培养形成单层后,100TCID50的病毒液和不超过20%细胞毒性的梯度浓度药物溶液同时加到MA104细胞上,继续培养4-6天后,更换含Alamarblue的培养液继续培养2~3小时后检测其530/590nm处的荧光值,并计算半数抑制浓度(IC50)。(c)根基TC50/IC50计算化合物的治疗指数。结果表明,本发明化合物的TC50值为218.5μg/mL、IC50值为6.48μg/mL,治疗指数TI为33.7;化合物具有很好的抗轮状病毒活性。以上结果揭示了本发明的化合物在制备抗轮状病毒药物中有良好的应用前景。本发明化合物结构简单活性较好,可作为抗轮状病毒药物研发的先导性化合物用于抗轮状病毒药物制剂研发。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1