1,3‑二取代环丁烷‑1,2,3,4‑四羧酸及其酸二酐的新型制造方法与流程
本发明涉及用于以高选择性且高收率获得1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐的新型制造方法。
背景技术:
1,3-二取代环丁烷-1,2,3,4-四羧酸二酐作为各种工业用途的原料是有用的。另外,成为所述1,3-二取代环丁烷-1,2,3,4-四羧酸二酐的原料的1,3-二取代环丁烷-1,2,3,4-四羧酸也是有用的化合物。
尤其是,式(12a)所示的1,3-二甲基环丁烷-1,2,3,4-四羧酸二酐和成为该酸二酐的原料的式(13a)所示的1,3-二甲基环丁烷-1,2,3,4-四羧酸是作为液晶表示元件、半导体中的保护材料、绝缘材料、滤色器、液晶取向膜、光波导用材料等而广泛使用的聚酰亚胺的主要原料或合成中间体(参照专利文献1和非专利文献1)。
作为式(12a)所示的1,3-二甲基环丁烷-1,2,3,4-四羧酸二酐和式(13a)所示的1,3-二甲基环丁烷-1,2,3,4-四羧酸的结构方面的特征,可列举出环丁烷环上的两个甲基位于1位和3位且该甲基的相对构型为反式。
以往,作为1,3-二取代环丁烷-1,2,3,4-四羧酸二酐的制造方法,已知下述制造方法。
根据非专利文献2,使柠康酸酐和作为光敏剂的二苯甲酮溶解于1,4-二噁烷,对该溶液照射光而进行环化反应,从而合成在环丁烷环上取代有两个甲基的二甲基环丁烷四羧酸二酐。进而,对所得二甲基环丁烷四羧酸二酐进行水解反应,接着,进行甲酯化反应,从而合成二甲基环丁烷-1,2,3,4-四羧酸四甲酯。但是,尚未确定环丁烷环上的甲基的位置和相对构型。
根据非专利文献3和非专利文献4,按照非专利文献2记载的方法,由柠康酸酐合成二甲基环丁烷-1,2,3,4-四羧酸二酐后,再合成二甲基环丁烷-1,2,3,4-四羧酸四甲酯。进而,记载了有可能生成由环丁烷环上的甲基位置和相对构型带来的4种二甲基环丁烷-1,2,3,4-四羧酸二酐的非对映体。
根据专利文献2,使柠康酸酐1000g溶解于醋酸乙酯,对该溶液照射光而进行环化反应,从而以695g的收量合成了柠康酸酐的二聚体。但是,针对所述柠康酸酐的二聚体、针对结构确定和非对映体的生成比没有任何记载。
根据专利文献3报告了:通过使乙基马来酸酐溶解于醋酸乙酯,并对该溶液照射光而进行环化反应,能够获得1,3-二乙基环丁烷-1,2,3,4-四羧酸1,2:3,4-二酐与1,2-二乙基环丁烷-1,2,3,4-四羧酸1,4:2,3-二酐的混合物。针对该混合物进行溶剂蒸馏去除、过滤、析晶等精制操作,但尚未实现彼此的分离精制。另外,通过收量和NMR算出的收率为中等程度。
现有技术文献
专利文献
专利文献1:国际公开(WO)2010/092989小册子
专利文献2:日本特开平4-106127号公报
专利文献3:日本特开2006-347931号公报
非专利文献
非专利文献1:新订最新聚酰亚胺基础和应用(日文:新訂最新ポリイミド基礎と応用)(ISBN 978-4-86043-273-7)、2010年、NTS Co.,Ltd.、344-354页
非专利文献2:化学学报(德文:Chemische Berichte)1962年、95卷、1642-1647页
非专利文献3:有机化学杂志(Journal of Organic Chemistry)1968年、33卷、920-921页
非专利文献4:化学学报(德文:Chemische Berichte)1988年、121卷、295-297页
技术实现要素:
发明要解决的问题
以往,尚未获知在构筑环丁烷环的反应时能够同时控制该环丁烷环上的取代基位置和相对构型的1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐的高选择性制造方法。因此,在获得目标化合物时,需要进行精制操作。具体而言,存在如下课题:需要利用升华来精制、使用大量有机溶剂进行析晶、利用悬浮清洗乃至过滤等进行复杂的精制操作,产生大量的废液、废弃物,从清洁化学的观点出发,对环境造成巨大的负担,生产率差。
本发明的目的在于,提供用于以高选择性和高收率获得1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐的新型制造方法,所述1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐作为各种工业用途的原料是有用,作为期望的立体结构,同时满足:环丁烷环上的两个取代基位于1位和3位,且该取代基的相对构型为反式。
用于解决问题的方案
本发明人为了解决上述课题而重复进行了深入研究,结果发现:通过生成由乙烯二羧酸衍生物和含氮有机化合物形成的晶体,并对该晶体照射光而进行环化反应,能够以高选择性和高收率获得具有期望立体结构的1,3-二取代环丁烷-1,2,3,4-四羧酸。本发明基于所述见解,具有下述主旨。
〔1〕用式(2)表示的1,3-二取代环丁烷-1,2,3,4-四羧酸的制造方法,其具有下述工序(a)和工序(b):
(R1表示C1~C4烷基、苯基或卤素原子。)
工序(a):在存在或不存在溶剂的条件下,制造由含氮有机化合物(5)与式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶体(1)的工序。
(R1表示与上述相同的意义。)
工序(b):对通过工序(a)得到的晶体(1)照射光而进行环化反应的工序。
〔2〕根据上述〔1〕所述的制造方法,其中,含氮有机化合物(5)为脂肪族胺、芳香族胺、氧化胺、酰胺、酰亚胺或含氮杂环式化合物。
〔3〕根据上述〔2〕所述的制造方法,其中,含氮有机化合物(5)为含氮杂环式化合物。
〔4〕根据上述〔3〕所述的制造方法,其中,含氮杂环式化合物为烟酰胺或吡啶。
〔5〕根据上述〔1〕~〔4〕中任一项所述的制造方法,其中,在工序(b)中,照射波长为290nm~600nm的光。
〔6〕根据上述〔1〕~〔4〕中任一项所述的制造方法,其中,在工序(b)中,照射波长为300nm~580nm的光。
〔7〕根据上述〔1〕~〔6〕中任一项所述的制造方法,其中,在工序(b)中,在光敏剂的存在下照射光。
〔8〕根据上述〔1〕~〔7〕中任一项所述的制造方法,其中,在式(4C)或式(4M)中,R1表示甲基或乙基。
〔9〕根据上述〔1〕~〔8〕中任一项所述的制造方法,其使用式(4C)所示的化合物。
〔10〕用式(3)表示的1,3-二取代环丁烷-1,2,3,4-四羧酸二酐的制造方法,其通过使利用上述〔1〕所述的制造方法得到的式(2)所示的1,3-二取代环丁烷-1,2,3,4-四羧酸发生脱水缩合反应来制造。
(R1表示C1~C4烷基、苯基或卤素原子。)
(式中,R1表示与上述相同的意义。)
〔11〕根据上述〔10〕所述的制造方法,其中,在醋酸酐的存在下进行脱水缩合反应。
〔12〕一种晶体,其由吡啶和柠康酸形成,所述晶体在基于Cu-Kα射线的粉末X射线衍射中在衍射角2θ=(12.58±0.2,15.05±0.2,16.08±0.2,17.60±0.2,19.20±0.2,21.57±0.2,23.02±0.2,24.50±0.2,26.45±0.2,27.06±0.2,28.10±0.2,32.49±0.2,35.90±0.2,36.46±0.2和38.43±0.2)处具有峰。
〔13〕根据上述〔12〕所述的晶体,其中,在基于Cu-Kα射线的粉末X射线衍射中在衍射角2θ=(12.58±0.2,15.05±0.2,16.08±0.2,17.60±0.2,18.34±0.2,19.20±0.2,21.57±0.2,23.02±0.2,24.50±0.2,26.45±0.2,27.06±0.2,28.10±0.2,30.39±0.2,32.49±0.2,34.71±0.2,35.90±0.2,36.46±0.2和38.43±0.2)处具有峰。
发明的效果
根据本发明,通过由乙烯二羧酸衍生物和含氮有机化合物制造晶体,接着对该晶体照射光而进行环化反应,能够以高选择性和高收率制造具有期望的立体结构的1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐。进而,本发明的制造方法在制造1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐时基本不产生副产物,因此,能够显著减轻为了获得具有期望的立体结构的化合物而进行的精制操作,因此,作为考虑了环境负担的工业制造方法是有用的。
附图说明
图1是实施例中制造的由吡啶和柠康酸形成的晶体(8)的FT-IR谱图。
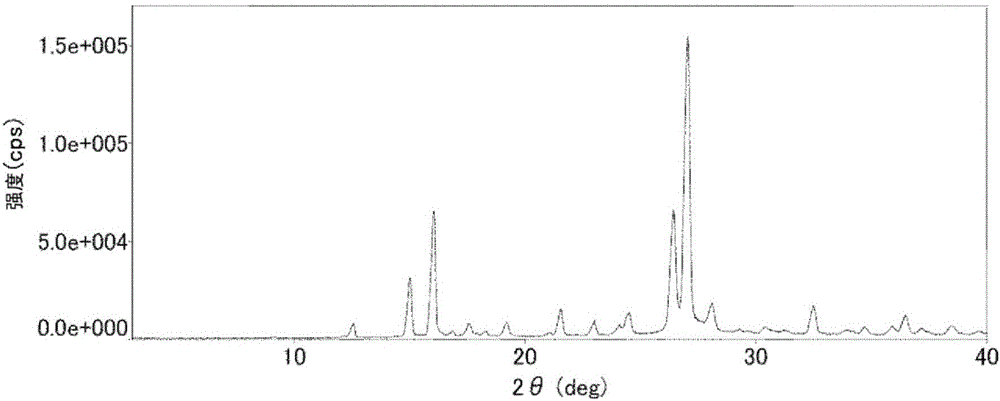
图2是实施例中制造的由吡啶和柠康酸形成的晶体(8)的粉末X射线衍射谱图。
图3是根据实施例中制造的晶体(8)的单晶X射线结构分析结果,利用圆柱模型一并示出吡啶嗡与柠康酸阴离子的堆积结构以及晶胞而得到的图。
图4是根据实施例中制造的晶体(8)的单晶X射线结构分析结果,利用ORTEP图仅示出柠康酸阴离子彼此的构型而得到的图。
图5是根据实施例中制造的化合物(13a)的单晶X射线结构分析结果,利用ORTEP图示出化合物(13a)的分子结构的图。
图6是根据实施例中制造的化合物(12a)的单晶X射线结构分析结果,利用ORTEP图示出化合物(12a)的分子结构的图。
图7是实施例9所使用的流动反应器的示意图。
图8是实施例9所使用的流动反应器中的具有双层管结构的T字型混合器(混合器2)的剖面图。
具体实施方式
本说明书中,下述符号分别表示下述意义。
“n-”表示正,“s-”表示仲,“t-”表示叔,“o-”表示邻,“m-”表示间,“p-”表示对。另外,“trans”、“cis”表示环状化合物的取代基的相对构型,该取代基位于环平面的相反侧时示为反式(trans),位于相同侧时示为顺式(cis)。另外,(E)和(Z)表示下述立体化学:用双键连结的分子的平面部分内键合于形成双键的原子上的基团之中,优先级规则的上位的基团存在于相反侧时示为(E),存在于同一侧时示为(Z)。
“Me”的表述是指甲基。Ca~Cb烷基表示从包含碳原子数a~b个的直链状或支链状的脂肪族烃中丧失1个氢原子而生成的一价基团。作为具体例,可列举出例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、叔戊基、1,1-二甲基丙基、正己基、异己基等。
作为卤素原子,可列举出氟原子、氯原子、溴原子、碘原子等。另外,卤素的表述也表示这些卤素原子。
以下,针对本发明中的1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐的制造方法、即工序(a)~工序(c)进行说明。
工序(a):制造由乙烯二羧酸衍生物和含氮有机化合物形成的晶体
本工序中,获得由含氮有机化合物(5)与式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶体(1)。式中,R1表示C1~C4烷基、苯基或卤素原子,其中,R1优选为甲基、乙基或苯基的情况,进一步优选为甲基或乙基的情况。
本说明书中的由含氮有机化合物(5)与式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶体(1)在室温下为固体,具有特征性的粉末X射线衍射峰,是由2种以上的化合物以一定的化学计量比形成的晶体。所述晶体(1)中,作为构成要素的2种以上的化合物在分子或离子层面上具有三维且周期性排列的结构。另外,晶体(1)由2种以上的成分构成,因此也可以说是所谓的多成分晶体。此处,室温是指1℃~40℃。
另一方面,所述晶体(1)中有时存在下述部分:晶体表面等处的周期性排列发生了崩塌的部分;乙烯二羧酸衍生物和含氮有机化合物在分子或离子层面上发生了聚集而不呈现周期结构的部分。但是,尽管局部具有三维周期性排列缺损的结构但包含上述三维且周期性排列的结构的晶体通过照射光而进行环化反应,从而能够高选择性地制造具有期望立体结构的1,3-二取代环丁烷-1,2,3,4-四羧酸时,也包括在本发明的晶体(1)中。
本发明中,工序(a)的由乙烯二羧酸衍生物和含氮有机化合物形成的晶体(1)的结构成为后续工序(b)中的1,3-二取代环丁烷-1,2,3,4-四羧酸、进而工序(c)中的1,3-二取代环丁烷-1,2,3,4-四羧酸二酐的立体选择性的决定性因素,因此,晶体(1)中的乙烯二羧酸衍生物的分子取向是重要的。本发明中的优选晶体(1)中,两个乙烯二羧酸衍生物以在后续工序的工序(b)的光环化反应中构筑两个甲基位于1位和3位且该甲基的相对构型呈现反式的环丁烷环的方式取向。
本发明中使用的属于乙烯二羧酸衍生物的物质是公知化合物,一部分可以以市售品的形式获取。例如,柠康酸可以由东京化成工业株式会社、Aldrich公司等获取。另外,苯基马来酸可以由HYDRUS CHEMICAL INC.获取。另外,中康酸可以由东京化成工业株式会社、Aldrich公司等来获取。
另外,本发明中使用的乙烯二羧酸衍生物的一部分可以按照文献记载的公知方法来合成。例如,2-异丙基马来酸可以参照合成通讯(Synthetic Communications)、2013年、43(10)卷、1455-1459页等来合成,苯基富马酸可以参照美国化学会志(Journal of the American Chemical Society)、1954年、76卷、1872-1873页等来合成。
作为本发明中使用的式(4C)所示的乙烯二羧酸衍生物的具体例,可列举出例如柠康酸、2-乙基马来酸、2-异丙基马来酸、2-丙基马来酸、2-正丁基马来酸、2-异丁基马来酸、2-(叔丁基)马来酸、2-苯基马来酸、2-氟马来酸、2-氯马来酸、2-溴马来酸、2-碘马来酸等。
其中,优选为柠康酸、2-乙基马来酸、2-异丙基马来酸或2-苯基马来酸,特别优选为柠康酸或2-乙基马来酸。
作为本发明中使用的式(4M)所示的乙烯二羧酸衍生物的具体例,可列举出例如中康酸、2-乙基富马酸、2-异丙基富马酸、2-丙基富马酸、2-正丁基富马酸、2-异丁基富马酸、2-(叔丁基)富马酸、2-苯基富马酸、2-氟富马酸、2-氯富马酸、2-溴富马酸、2-碘富马酸等。
其中,优选为中康酸、2-乙基富马酸、2-苯基富马酸或2-氟富马酸,特别优选为中康酸或2-乙基富马酸。
作为含氮有机化合物(5),可以使用各种化合物。例如可列举出:在生成上述晶体(1)时,通过在晶体结构中在酸性的式(4C)或式(4M)所示的乙烯二羧酸衍生物与含氮有机化合物(5)之间发生质子移动,从而借助所形成的离子键来生成晶体的例子。因此,可以使用能够与酸性的乙烯二羧酸衍生物发生中和反应那样的具有碱性的多种含氮有机化合物。
另外,可以使用具有如下取代基、部分骨架的多种含氮有机化合物,所述取代基、部分骨架在生成晶体(1)时,能够与乙烯二羧酸衍生物形成氢键、范德华力等分子间相互作用。作为该取代基,可列举出氨基、酰胺基、亚氨基、醚基、羟基、羰基、羧基等,作为该部分骨架,可列举出吡咯系骨架、吡咯啉系骨架、吡咯烷系骨架、吲哚系骨架、吲哚啉系骨架、异吲哚系骨架、咪唑啉系骨架、咪唑烷系骨架、吡啶系骨架、哌啶系骨架、喹啉系骨架、吖啶系骨架、三嗪系骨架等。
本发明中,在后续的工序(b)中,对于由含氮有机化合物(5)与式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶体(1)中的乙烯二羧酸衍生物的双键照射光而使其进行环化反应,因此,作为含氮有机化合物,优选不阻碍光环化反应的推进。因此优选为下述化合物:不与乙烯二羧酸衍生物反应而产生副产物的化合物、具有在生成目标化合物之前不易进行晶体(1)被破坏之类的异构化反应等的结构的化合物、具有耐光照射性的结构的化合物等。作为所述含氮有机化合物的优选例,可列举出脂肪族胺、芳香族胺、氧化胺、酰胺、酰亚胺、含氮杂环式化合物等。
作为上述脂肪族胺,可列举出甲胺、乙胺、异丙胺、丁胺、异丁胺、仲丁胺、叔丁胺、戊胺、异戊胺、2-戊胺、叔戊胺、己胺、庚胺、辛胺、2-辛胺、2-乙基己胺、壬胺、癸胺、苄胺、苯乙胺、α-甲基苄胺、墨斯卡灵、多巴胺、二丙胺、二异丙胺、N-甲基乙胺、N-乙基异丁胺、二丁胺、二异丁胺、二苄胺、N,N-二甲基丙胺、三丙胺、N-乙基-N-甲基丁胺、三丁胺、N,N-二甲基苄胺、N,N-二乙基苄胺、三苄胺、环丙胺、环丁胺、环己胺、二环己胺、N,N-二甲基环己胺、环己烷-1β,2β-二胺、(1R,2R)-1,2-环己烷二胺、(1S,2S)-1,2-环己烷二胺等。其中,优选为甲胺、乙胺、环己烷-1β,2β-二胺、(1R,2R)-1,2-环己烷二胺、或者(1S,2S)-1,2-环己烷二胺。
作为上述芳香族胺,可列举出邻甲苯胺、间甲苯胺、对甲苯胺、邻乙基苯胺、间乙基苯胺、对乙基苯胺、对异丙基苯胺、对叔戊基苯胺、二甲苯胺、2,3-二甲苯胺、2,4-二甲苯胺、2,6-二甲苯胺、3,4-二甲苯胺、3,5-二甲苯胺、百里基胺、2,4,5-三甲基苯胺、2,4,6-三甲基苯胺、五甲基苯胺、1-萘胺、2-萘胺、1-蒽基胺、2-蒽基胺、9-蒽基胺、N-丁基苯胺、N-异戊基苯胺、N-苄基苯胺、N-苄基-N-乙基苯胺、N,N-二苯基苄胺、N-甲基邻甲苯胺、N-甲基对甲苯胺、N,N-二甲基苯胺、N,N-二乙基苯胺、N,N-二丁基苯胺、N,N-二戊基苯胺、N,N-甲基邻甲苯胺、N,N-甲基间甲苯胺、N,N-甲基对甲苯胺、二苯基胺、二对甲苯胺、N-甲基二苯基胺、N-乙基二苯基胺、三苯基胺、N,N-二苄基苯胺、N-苄基-N-乙基苯胺、邻苯二胺、间苯二胺、对苯二胺等。其中,优选为邻甲苯胺、间甲苯胺、对甲苯胺、邻乙基苯胺、间乙基苯胺或对乙基苯胺。
作为上述氧化胺,可列举出三甲基氧化胺、吡啶1-氧化物、2,2’-联吡啶1,1’-二氧化物、4,4’-二甲基-2,2’-联吡啶1,1’-二氧化物、3,3’-二甲基-2,2’-联吡啶1,1’-二氧化物等。其中,优选为三甲基氧化胺、吡啶1-氧化物或3,3’-二甲基-2,2’-联吡啶1,1’-二氧化物。
作为上述酰胺,可列举出甲酰胺、N,N-二甲基甲酰胺、N,N-二异丙基甲酰胺、乙酰胺、N-乙基乙酰胺、N,N-二甲基乙酰胺、N-氯乙酰胺、N-溴乙酰胺、二乙酰胺、三乙酰胺、丙酰胺、丁酰胺、异丁酰胺、戊酰胺、异戊酰胺、己酰胺、庚酰胺、辛酰胺、丙烯酰胺、氯乙酰胺、二氯乙酰胺、三氯乙酰胺、羟基乙酰胺、乳酰胺、丙酮酰胺、氰基乙酰胺、雷尿酸(fulminuric acid)、草酰胺、丙二酰胺、琥珀酰胺、己二酰胺、L-苹果酰胺、(R,R)-酒石酰胺等。其中,优选为甲酰胺、N,N-二甲基甲酰胺、N,N-二异丙基甲酰胺或乙酰胺。
作为上述酰亚胺,可列举出琥珀酰亚胺、N-溴代琥珀酰亚胺、N-氯代琥珀酰亚胺等。其中,优选为琥珀酰亚胺。
作为上述含氮杂环式化合物,可列举出吡咯系化合物、吡咯啉系化合物、吡咯烷系化合物、吲哚系化合物、吲哚啉系化合物、异吲哚系化合物、咔唑系化合物、二唑系化合物、咪唑啉系化合物、咪唑烷系化合物、吡啶系化合物、吡啶的取代衍生物、哌啶系化合物、喹啉系化合物、氢喹啉系化合物、异喹啉系化合物、吖啶系化合物、菲啶系化合物、二嗪系化合物、磺胺嘧啶系化合物、氢嘧啶系化合物、哌嗪系化合物、苯并二嗪系化合物、三唑系化合物、苯并三唑系化合物、三嗪系化合物、嘌呤、次黄嘌呤、黄嘌呤、可可碱、茶碱、咖啡因、尿酸、腺嘌呤、鸟嘌呤、3-甲基尿酸、7-甲基尿酸等。
作为上述吡咯系化合物,可列举出吡咯、甲基吡咯、二甲基吡咯、3-乙基-4甲基吡咯、乙基二甲基吡咯、3-乙基-2,4,5-三甲基吡咯、2,3,4,5-四甲基吡咯、乙酰基吡咯等。作为上述吡咯啉系化合物,可列举出吡咯啉等。作为上述吡咯烷系化合物,可列举出吡咯烷等。作为上述吲哚系化合物,可列举出吲哚、假吲哚、甲基吲哚、2,3-二甲基吲哚、2-苯基吲哚等。作为上述吲哚啉系化合物,可列举出吲哚啉、靛红、O-甲基靛红、N-甲基靛红、2-氯-3-吲哚酮等。
作为上述异吲哚系化合物,可列举出异吲哚、异吲哚啉、苯并吡咯酮等。作为上述咔唑系化合物,可列举出咔唑、靛蓝、靛白(leucoindigo)、靛玉红等。作为上述二唑系化合物,可列举出吡唑、3,5-二甲基吡唑、2-吡唑啉、吡唑烷、吡唑啉酮、3-甲基-1-苯基-5-吡唑啉酮、2,3-二甲基-1-苯基-5-吡唑啉酮、氨基比林、吲唑等。作为上述咪唑啉系化合物,可列举出咪唑啉、苦杏素(amarine)、萘甲唑啉等。作为上述咪唑烷系化合物,可列举出乙烯脲、乙内酰脲、1-甲基乙内酰脲、5-甲基乙内酰脲、二苯基乙内酰脲、肌酸酐等。
作为上述吡啶系化合物,可列举出吡啶、2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-乙基吡啶、3-乙基吡啶、4-乙基吡啶、2-丙基吡啶、2,3-二甲基吡啶、2,4-二甲基吡啶、2,5-二甲基吡啶、2,6-二甲基吡啶、3,4-二甲基吡啶、3,5-二甲基吡啶、4-乙基-2-甲基吡啶、3-乙基-4-甲基吡啶、5-乙基-2-甲基吡啶、6-乙基-2-甲基吡啶、2,4,6-三甲基吡啶、2,3,4-三甲基吡啶、4-乙基-2,6-二甲基吡啶、2-苯基吡啶、3-苯基吡啶、4-苯基吡啶、2-苄基吡啶、3-苄基吡啶、4-苄基吡啶、2,2’-联吡啶、3,3’-联吡啶、4,4’-联吡啶等。
作为上述吡啶的取代衍生物,可列举出2-氯吡啶、3-氯吡啶、4-氯吡啶、2-吡啶酮、3-吡啶酚、4-吡啶酮、2-甲氧基吡啶、3-甲氧基吡啶、4-甲氧基吡啶、2-吡啶甲醛、3-吡啶甲醛、4-吡啶甲醛、2-乙酰基吡啶、3-乙酰基吡啶、4-乙酰基吡啶、2-乙氧基吡啶1-氧化物、2-吡啶羧酸、烟酸、异烟酸、烟酰胺、尼可刹米、异烟肼、2-乙基硫异烟胺、2,3-吡啶二羧酸、2,4-吡啶二羧酸、2,5-吡啶二羧酸、2,6-吡啶二羧酸、3,4-吡啶二羧酸、3,5-吡啶二羧酸、3-硝基吡啶、2-吡啶胺、3-吡啶胺、4-吡啶胺、N,N-二甲基-4-吡啶胺等。
作为上述哌啶系化合物,可列举出哌啶、2-甲基哌啶、毒芹碱(coniine)、3-甲基哌啶、4-甲基哌啶、N-甲基哌啶、1-苯基哌啶、2,6-二甲基哌啶、N-苯甲酰基哌啶、4-哌啶酮等。作为上述喹啉系化合物,可列举出喹啉、2-甲基喹啉、3-甲基喹啉、4-甲基喹啉、6-甲基喹啉、8-甲基喹啉、2,3-二甲基喹啉、2,4-二甲基喹啉、2,6-二甲基喹啉、2-苯基喹啉、6-苯基喹啉、8-苯基喹啉、2-氯喹啉、2-喹诺酮、4-喹诺酮、5-喹啉醇、6-喹啉醇、7-喹啉醇、8-喹啉醇、α-萘喹啉、β-萘喹啉、2-甲基-4-喹啉醇、4-甲基-2-喹啉醇、6-甲氧基喹啉、2,4-喹啉二醇、2-喹啉羧酸、3-喹啉羧酸、4-喹啉羧酸、5-喹啉羧酸、5-硝基喹啉、6-硝基喹啉、7-硝基喹啉、8-硝基喹啉、2-喹啉胺、3-喹啉胺、4-喹啉胺、黄苯胺、2,2’-二喹啉、3,3’-二喹啉、5,5’-二喹啉、6,6’-二喹啉、2,3’-二喹啉等。
作为上述氢喹啉系化合物,可列举出1,2,3,4-四氢喹啉、1-甲基-1,2,3,4-四氢喹啉、6-甲氧基-1,2,3,4-四氢喹啉、3,4-二氢-2-喹诺酮、顺式-十氢喹啉等。作为上述异喹啉系化合物,可列举出异喹啉、1-甲基异喹啉、1-异喹诺酮、1,2,3,4-四氢异喹诺酮、1-苄基异喹啉等。作为上述吖啶系化合物,可列举出吖啶、2-甲基吖啶、3-甲基吖啶、9-甲基吖啶、9-苯基吖啶、3-氨基-9-(对氨基苯基)吖啶、3,6-二氨基-10-甲基氯化吖啶、吖啶满、吖啶酮、吖啶醇等。
作为上述菲啶系化合物,可列举出菲啶、苯并[f]喹啉、苯并[g]喹啉、苯并[h]喹啉、苯并[g]异喹啉、1,10-菲咯啉、2,9-二甲基-1,10-菲咯啉、4,7-二苯基-1,10-菲咯啉、2,9-二甲基-4,7-二苯基-1,10-菲咯啉等。作为上述二嗪系化合物,可列举出哒嗪、嘧啶、吡嗪、2,5-二甲基吡嗪、四苯基吡嗪等。
作为上述磺胺嘧啶系化合物,可列举出磺胺嘧啶、碘胺二甲氧哒嗪、磺胺苯吡唑、磺胺甲噻二唑、磺胺甲氧基哒嗪、磺胺异噁唑、磺胺索嘧啶等。作为上述氢嘧啶系化合物,可列举出尿嘧啶、胸腺嘧啶、扑米酮、2-硫尿嘧啶、胞嘧啶、巴比妥酸、5,5-二乙基巴比妥酸、5,5-二丙基巴比妥酸、阿洛巴比妥、5-乙基-5-苯基巴比妥酸、环巴比妥(cyclobarbital)、环己烯巴比妥(hexobarbital)、5-羟基巴比妥酸、5-硝基巴比妥酸、5-氨基巴比妥酸、异戊巴比妥、紫尿酸、阿脲、双阿脲、红紫酸、紫螺酸铵、苦杏酸等。作为上述哌嗪系化合物,可列举出哌嗪、2,5-二甲基哌嗪、2,5-哌嗪二酮等。作为上述苯并二嗪系化合物,可列举出噌啉、酞嗪、喹唑啉、喹喔啉等。作为上述三唑系化合物,可列举出1,2,3-三唑、1,2,4-三唑、4-氨基-1,2,4-三唑等。
作为上述苯并三唑系化合物,可列举出苯并三唑、5-甲基苯并三唑等。作为上述三嗪系化合物,可列举出1,2,3-三嗪、4-甲基-1,2,3-三嗪、4,6-二甲基-1,2,3-三嗪、4,5,6-三甲基-1,2,3-三嗪、1,2,3-苯并三嗪、4-甲基-1,2,3-苯并三嗪、1,3,5-三嗪、氰尿酰氯、氰尿酸、氰尿酸三甲酯、异氰尿酸甲酯、异氰尿酸乙酯、黑色素、氰尿二酰胺、氰尿酰胺等。
本发明中,作为优选的含氮杂环式化合物,可列举出吡啶系化合物、吡啶的取代衍生物。作为优选的吡啶系化合物,可列举出吡啶、2,3-二甲基吡啶、3,5-二甲基吡啶,作为更优选的吡啶系化合物,可列举出吡啶。作为优选的吡啶的取代衍生物,可列举出烟酸、异烟酸、烟酰胺、尼可刹米,作为更优选的吡啶的取代衍生物,可列举出烟酰胺。
上述含氮有机化合物可以单独使用,也可以组合使用这些之中的2种以上。
本发明中,由含氮有机化合物(5)与式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶体(1)可通过将两者混合而使其充分接触来制造。关于制造晶体(1)时的含氮有机化合物的用量,相对于乙烯二羧酸衍生物1当量,含氮有机化合物优选为0.1~50当量、更优选为0.2~15当量、特别优选为0.5~3当量。
尤其是,含氮有机化合物为液体时,也可以用作乙烯二羧酸衍生物的溶剂,乙烯二羧酸衍生物为液体时,也可以用作含氮有机化合物的溶剂。
制造晶体(1)时的温度没有特别限定,可以在-78℃~反应混合物的回流温度内任意设定。其中,优选为-30~70℃,更优选为-20~40℃。
制造晶体(1)时的压力可以为加压、常压或减压中的任一者,优选为0.5~10atm,更优选为0.9~2atm。
制造晶体(1)时的将式(4C)或式(4M)所示的乙烯二羧酸衍生物与含氮有机化合物(5)进行混合的方法可以使用如下方法:将两者直接混合的方法、使用溶剂进行混合的方法等。
作为将两者直接混合的方法,可以使用捏合法、混合粉碎法等。捏合法可应用于一者为固体且另一者为液体时的混合,在少量的情况下可以使用乳钵,在大量的情况下可以使用捏合机、有机合成用的反应容器、搅拌机等。
乙烯二羧酸衍生物和含氮有机化合物均为固体时,可以应用混合粉碎法,在少量的情况下可以使用乳钵,在大量的情况下可以使用粉碎机等。利用捏合法或混合粉碎法进行混合时,有时因中和反应等而伴有发热,因此,也可以一边对乳钵、反应容器等进行冷却一边实施。
作为将乙烯二羧酸衍生物和含氮有机化合物用溶剂进行混合的方法,可以采取使用了溶剂的温度控制法、溶剂蒸发法、溶剂蒸馏去除法、不良溶剂添加法、溶液-溶液混合法、悬浮液-溶液混合法、悬浮液-悬浮液混合法等。另外,这些混合时,有时因溶解、中和反应等而伴有发热,因此,也可以一边对反应容器等进行冷却一边实施。
通过上述使用了溶剂的温度控制法来制造晶体(1)的方法是如下方法:首先,使乙烯二羧酸衍生物与含氮有机化合物的反应混合物预先溶解于溶剂,利用高温与低温的溶解度差来获得晶体的方法。
通过上述使用了溶剂的溶剂蒸发法或溶剂蒸馏去除法来制造晶体(1)的方法是首先使乙烯二羧酸衍生物与含氮有机化合物的反应混合物溶解于溶剂,并使溶剂蒸发或蒸馏去除的方法,在使其缓慢蒸发的情况下,大多能够获得良好的晶体。蒸馏去除溶剂时,可以使用旋转蒸发仪等。无法利用1种溶剂良好地进行结晶化时,也可以使用混合溶剂。
通过使用了溶剂的不良溶剂添加法来制造晶体(1)的方法是首先使乙烯二羧酸衍生物与含氮有机化合物的反应混合物溶解于溶剂,并通过添加不良溶剂来获得晶体的方法。根据所用溶剂的不同,有时乙烯二羧酸衍生物与含氮有机化合物的反应混合物的一部分不会溶解,从而呈现在液体中分散有固体颗粒的浆料。
针对通过使用了溶剂的溶液和悬浮液来制造晶体(1)的方法,以下进行说明。首先,使乙烯二羧酸衍生物溶解于溶剂来制备溶液A。同样制备含氮有机化合物的溶液B。进而,制备使乙烯二羧酸衍生物悬浮于溶剂的悬浮液A,同样制备含氮有机化合物的悬浮液B。由上述溶液和悬浮液分别选择乙烯二羧酸衍生物和含氮有机化合物的溶液或悬浮液并混合,从而得到晶体的方法。如下述那样,混合有如下方法:同时滴加液体的方法、顺序滴加的方法、或者反序添加的方法等。此时使用的溶剂可以仅为1种,也可以利用将多种溶剂混合得到的溶剂。
乙烯二羧酸衍生物的溶液A和悬浮液A的浓度只要不阻碍生成晶体(1)的反应就没有特别限定,优选为0.01~100mol/L、更优选为0.05~10mol/L、特别优选为0.2~3mol/L。
含氮有机化合物的溶液B和悬浮液B的浓度只要不阻碍生成晶体(1)的反应就没有特别限定,优选为0.01~100mol/L、更优选为0.05~10mol/L、特别优选为0.2~3mol/L。
溶液-溶液混合法是通过将上述溶液A与溶液B混合而得到晶体(1)的方法。此时的混合中,将同时滴加溶液A和溶液B的方法记作同时滴加,将向溶液B中滴加溶液A的方法记作顺序滴加,将向溶液A中滴加溶液B的方法记作反序滴加。
悬浮液-溶液混合法是通过将上述溶液A与悬浮液B混合、或者将悬浮液A与溶液B混合而得到晶体(1)的方法。此时的混合可以从同时滴加、顺序滴加或反序滴加中选择。
悬浮液-悬浮液混合法是通过将上述悬浮液A与悬浮液B混合而得到晶体(1)的方法。此时的混合可以从同时滴加、顺序滴加或反序滴加中选择。
本发明中,也可以按照任意的顺序将上述方法组合实施。例如,可以向该不良溶剂添加法中按照任意的顺序组合溶剂蒸馏去除法、使用了溶剂的温度控制法来实施,能够构筑用于制造晶体(1)的条件。
使用了溶剂的晶体(1)的制造方法中,溶液或悬浮液的搅拌效率高是较好的。以下说明此时能够使用的搅拌装置,但不限定于下述记载。作为装置,可列举出捏合机、有机合成用的反应容器、搅拌机、超声波发生器、流动反应器的混合器等。具体而言,可列举出基于磁力搅拌棒和磁力搅拌器的搅拌、基于搅拌叶片的搅拌、基于非活性气体鼓泡的搅拌、基于离心式搅拌体的搅拌、能够配置在反应容器中的挡板等。针对搅拌叶片,优选为螺旋叶片、桨式叶片、MAXBLEND叶片(注册商标)、盘式涡轮叶片、FULLZONE叶片(注册商标)等,优选为MAXBLEND叶片(注册商标)、盘式涡轮叶片或FULLZONE叶片(注册商标)。
利用工序(a)制造的晶体(1)还因其性状而异,可以通过过滤等来进行分离,也可以不分离地连续实施后续工序(b)中的照射光的环化反应。本说明书中,将后者称为工序(a)和工序(b)的连续化。
使用溶剂将上述乙烯二羧酸衍生物与含氮有机化合物进行混合的方法中使用的溶剂只要不阻碍生成晶体(1)的反应就没有特别限定。作为所述混合时使用的溶剂,可列举出例如甲苯、邻二甲苯等芳香族烃系溶剂、己烷、庚烷、石油醚等脂肪族烃系溶剂、环己烷等脂环式烃系溶剂、氯苯、邻二氯苯等芳香族卤代烃系溶剂、二氯甲烷、氯仿、四氯化碳、1,2-二氯乙烷、1,1,1-三氯乙烷、三氯乙烯、四氯乙烯等脂肪族卤代烃系溶剂、二乙基醚、二异丙基醚、1,2-二甲氧基乙烷、四氢呋喃、1,4-二噁烷、环戊基甲基醚等醚系溶剂、三乙基胺、三丁基胺、N,N-二甲基苯胺等胺系溶剂、吡啶、甲基吡啶等吡啶系溶剂、醋酸乙酯、醋酸正丁酯、丙酸乙酯等酯系溶剂、甲醇、乙醇、正丙醇、2-丙醇、乙二醇等醇系溶剂、丙酮、甲基异丁基酮等酮系溶剂、碳酸二甲酯、碳酸二乙酯等碳酸酯系溶剂、乙腈、二甲基亚砜、环丁砜、1,3-二甲基-2-咪唑烷酮、乙二醇二乙酸酯、醋酸、水等。
其中,作为优选的溶剂,可列举出甲苯、邻二甲苯、己烷、庚烷、石油醚、邻二氯苯、二氯甲烷、二乙基醚、四氢呋喃、1,4-二噁烷、吡啶、醋酸乙酯、醋酸正丁酯、甲醇、乙醇、2-丙醇、丙酮、甲基异丁基酮、碳酸二甲酯、碳酸二乙酯、乙腈、乙二醇二乙酸酯或醋酸。作为更优选的溶剂,可列举出己烷、庚烷、四氢呋喃、吡啶、醋酸乙酯、醋酸正丁酯、甲醇、乙醇、碳酸二甲酯或醋酸。这些溶剂可以单独使用,或者混合使用2种以上。
作为利用工序(a)制造的晶体(1)的代表例,将通过后述实施例1得到的由吡啶和柠康酸形成的晶体(8)在使用Cu-Kα辐射线的粉末X射线衍射中显示峰值的衍射角2θ的值(也简称为峰值)示于〔表1〕。
〔表1〕
〔表1〕记载的晶体(8)的峰值是根据下述实施例1记载的方法得到的3个批次的晶体(8)的峰值的平均值。
应予说明,粉末X射线衍射的任意峰通常均存在±0.2的误差。将考虑了该误差值的晶体(8)的峰值示于〔表2〕。
〔表2〕
接着,将晶体(8)的粉末X射线衍射峰之中的特别的特征性峰值示于〔表3〕。
〔表3〕
将考虑了误差值的晶体(8)的特征性峰值示于〔表4〕。
〔表4〕
工序(b):对由乙烯二羧酸衍生物和含氮有机化合物形成的晶体(1)照射光来进行环化反应,从而制造1,3-二取代环丁烷-1,2,3,4-四羧酸
本工序(b)中,通过对由乙烯二羧酸衍生物和含氮有机化合物形成的晶体(1)照射光,针对晶体中的乙烯二羧酸衍生物的双键进行环化反应,从而制造式(2)所示的1,3-二取代环丁烷-1,2,3,4-四羧酸。式中的R1如上所示。
本发明的照射光的环化反应的特征在于,其是在晶体中、即固相中的反应,起始原料为固体状态,因此在原子、分子的移动显著受限的环境下发生。
作为本发明中的对晶体(1)照射光的环化反应的光源的波长,优选为280~600nm、更优选为290~600nm、进一步优选为300~580nm。
作为光源,可列举出高压汞灯、低压汞灯、超高压汞灯、氙气灯、钠灯、卤素灯、气体激光、液体激光、固体激光、太阳光、发光二极管等。其中,优选为高压汞灯或发光二极管,特别优选为高压汞灯。
光源为了冷却而优选容纳于夹套,作为此时的夹套材质,可列举出石英玻璃、Pyrex(注册商标)、HARIO玻璃、钼玻璃、钠玻璃、铅玻璃、钨玻璃、VYCOR(注册商标)、合成石英玻璃(SUPRASIL)等。作为优选的夹套材质,为石英玻璃、Pyrex、HARIO玻璃、钼玻璃等,作为更优选的夹套材质,为Pyrex。
作为本发明中的对晶体(1)照射光的环化反应中的光源照射方法,可以使用例如将光源设置于反应器内侧的内部照射、设置于反应器外侧的外部照射等。
作为具体的光源照射方法,有直接对晶体(1)进行光照射的方法、对使晶体(1)分散在溶剂中而得到的浆料照射光的方法等。对浆料照射光时,可以使用将光源设置于反应容器内部的内部照射以及设置于反应容器外侧的外部照射中的任一者。
对晶体(1)照射光的环化反应中的气氛可列举出大气、通常的空气、氮气、氦气、氩气等。其中,优选为大气、空气、氮气或氩气,特别优选为大气或氮气。
对晶体(1)照射光而实施环化反应时的反应温度没有特别限定,可以设定为-78℃~反应混合物的回流温度。其中,优选为-40~90℃,更优选为-20~50℃。
对晶体(1)照射光而实施环化反应时的反应装置没有特别限定,从形状出发,可以使用槽型、管型等,从操作方法出发,可以使用间歇式、连续式、半间歇式等。可列举出例如间歇反应器、连续槽型反应器、活塞流反应器、流动反应器等。
工序(b)中,对使晶体(1)分散在溶剂中而得到的浆料照射光时,该浆料的搅拌效率优选较高。以下说明此时可以使用的搅拌装置,但不限定于下述装置。作为装置,可列举出有机合成用的反应容器、搅拌机、超声波发生器、流动反应器的混合器等。具体而言,可列举出基于磁力搅拌棒和磁力搅拌器的搅拌、基于搅拌叶片的搅拌、基于非活性气体鼓泡的搅拌、基于离心式搅拌体的搅拌、能够配置在反应容器中的挡板等。针对搅拌叶片,优选为螺旋叶片、桨式叶片、MAXBLEND叶片(注册商标)、盘式涡轮叶片、FULLZONE叶片(注册商标)等,优选为对于浆料的搅拌效率良好的搅拌叶片,更优选为MAXBLEND叶片(注册商标)、盘式涡轮叶片或FULLZONE叶片(注册商标)。
本发明中的对晶体(1)照射光的环化反应也可以在光敏剂的存在下进行。作为光敏剂,可列举出例如苯、甲苯、丙酮、丁烷-2,3-二酮、杜烯、苯甲腈、苯丁酮、苯丙酮、苯乙酮、氧杂蒽酮、4-甲氧基苯乙酮、4’-乙酰基苯乙酮、蒽酮、苯甲醛、4,4’-二甲氧基二苯甲酮、二苯甲酮、芴、苯并菲、联苯、硫杂蒽酮、蒽醌、4,4’-双(二甲氨基)二苯甲酮、菲、萘、4-苯基苯乙酮、4-苯基二苯甲酮、2-碘萘、1,2-二脱氢苊烯(1,2-didehydroacenaphthylene)、2-萘甲腈、1-碘萘、1-萘甲腈、屈、晕苯、苯偶酰、荧蒽、芘、1,2-苯并蒽、吖啶、蒽、苝、萘并萘、2-甲氧基萘、2-乙酰基萘、1,4’-二氰基萘、9-氰基蒽、9,10-二氰基蒽、9,10-二溴蒽、2,6,9,10-四氰基蒽等。
其中,光敏剂优选为苯、甲苯、丙酮、丁烷-2,3-二酮、苯甲腈、苯丁酮、苯丙酮、苯乙酮、氧杂蒽酮、4-甲氧基苯乙酮、4’-乙酰基苯乙酮、蒽酮、苯甲醛、4,4’-二甲氧基二苯甲酮、二苯甲酮、芴、苯并菲、联苯、硫杂蒽酮、蒽醌、4,4’-双(二甲基氨基)二苯甲酮、菲、萘、4-苯基苯乙酮、4-苯基二苯甲酮、1,2-二脱氢苊烯、苯偶酰、芘、吖啶、蒽、苝、2-乙酰基萘或9,10-二溴蒽。
作为本发明中的对晶体(1)照射光的环化反应中的用于将晶体(1)制成浆料的溶剂,可列举出例如甲苯、邻二甲苯等芳香族烃系溶剂、己烷、庚烷、2-甲基戊烷、辛烷、石油醚等脂肪族烃系溶剂、环己烷等脂环式烃系溶剂、氯苯、邻二氯苯等芳香族卤代烃系溶剂、二氯甲烷、氯仿、四氯化碳、1,2-二氯乙烷、1,1,1-三氯乙烷、三氯乙烯、四氯乙烯等脂肪族卤代烃系溶剂、二乙基醚、二异丙基醚、1,2-二甲氧基乙烷、四氢呋喃、1,4-二噁烷、环戊基甲基醚等醚系溶剂、三乙基胺、三丁基胺、N,N-二甲基苯胺等胺系溶剂、吡啶、甲基吡啶等吡啶系溶剂、醋酸乙酯、醋酸正丁酯、丙酸乙酯等酯系溶剂、甲醇、乙醇、正丙醇、2-丙醇、正丁醇、2-丁醇、异丁醇、叔丁醇、1-戊醇、2-戊醇、3-戊醇、1-辛醇、2-辛醇、乙二醇等醇系溶剂、丙酮、甲基异丁基酮等酮系溶剂、碳酸二甲酯、碳酸二乙酯等碳酸酯系溶剂、乙腈、二甲基亚砜、环丁砜、1,3-二甲基-2-咪唑烷酮、乙二醇二乙酸酯、醋酸、醋酸酐、水等。
其中,用于将晶体(1)制成浆料的溶剂优选为甲苯、邻二甲苯、己烷、庚烷、石油醚、邻二氯苯、二乙基醚、二异丙基醚、吡啶、醋酸乙酯、醋酸正丁酯、甲基异丁基酮或碳酸二甲酯。这些溶剂可以单独使用,也可以混合使用2种以上。
本发明中,将晶体(1)制成浆料并照射光来进行环化反应的优点可列举出:能够对分散在溶液中的晶体高效地照射光、可容易地控制光反应实施中的反应温度等。此时,作为制备晶体(1)的浆料时的浓度,优选为0.001~100mol/L、更优选为0.01~10mol/L、特别优选为0.05~3mol/L。
工序(c):1,3-二取代环丁烷-1,2,3,4-四羧酸二酐的制造
工序(c)中,以式(2)所示的1,3-二取代环丁烷-1,2,3,4-四羧酸作为起始原料,通过脱水缩合反应来制造式(3)所示的1,3-二取代环丁烷-1,2,3,4-四羧酸二酐。式中的R1如上所示。
所述工序(c)的反应可根据已知的方法、例如合成通讯(Synthetic Communications)、1989年、19(3-4)卷、679-688页;合成通讯(Synthetic Communications)、1987年、17(3)卷、355-368页;合成通讯(Synthetic Communications)、2003年、33(8)卷、1275-1283页等记载的方法来实施。
作为本发明的工序(c)的脱水缩合反应中使用的缩合剂,可列举出例如醋酸酐、亚硫酰氯、乙酰氯等。另外,缩合剂也可以用作溶剂。
作为工序(c)的脱水缩合反应时使用的溶剂,只要不阻碍反应的进行就没有特别限定,可列举出例如甲苯、醋酸乙酯、醋酸酐、亚硫酰氯、乙酰氯、吡啶等。这些溶剂可以单独使用,也可以混合使用2种以上。另外,也可以在无溶剂的条件下实施该脱水缩合反应。
工序(c)的脱水缩合反应的反应温度可以设定为-60℃~反应混合物的回流温度之间的任意温度。其中,优选为-20~230℃,更优选为0~150℃。
工序(c)在无溶剂的条件下进行的脱水缩合反应的反应温度可以设定为100℃~起始原料的热分解温度之间的任意温度。反应温度优选为100~230℃。
实施例
以下,通过本发明的实施例来具体说明,但本发明不限定于它们。实施例中使用的分析装置、光反应用光源如下所示。
NMR:
ECX 300(JEOL公司制):1H-NMR、13C-NMR
化学位移值通过使用Me4Si(四甲基硅烷)作为内标物质,并利用氘代二甲基亚砜(DMSO-d6)溶剂来测定。
JNM-ECA500(JEOL公司制):定量1H-NMR(13C去耦1H测定)
作为标准物质使用马来酸,用氘代二甲基亚砜(DMSO-d6)溶剂进行测定。
AVANCE III 500(Bruker公司制):固相13C-NMR
作为标准物质使用金刚烷来进行测定。
气相色谱法(GC):
GC:6890series GC(Hewlett Packard公司制)
气相色谱-高分辨率质谱(GC-HRMS):
GC:7890A(Agilent公司制),MS:GCT Premier(Waters公司制)
单晶X射线结构分析:
SMART APEX II ULTRA(Bruker公司制)
粉末X射线衍射:
MiniFlex600(Rigaku公司制)
红外吸收(IR):
FT-IR ALPHA(Bruker Optics公司制)
光源:作为100W高压汞灯,使用SEN LIGHTS Co.,Ltd.制造的电源:HB100P-1(5/6)、光源:HL100CH-4,作为400W高压汞灯,使用SEN LIGHTS Co.,Ltd.制造的电源:HB400P-1、光源:HL400B(L/H/S)-8,作为450W高压汞灯,使用USHIO INC.制造的UM-452。作为氙气灯,使用USHIO INC.制造的UV-XEFL(主波长峰为290nm、4.5W〔使用1.5W/cm的长度3cm的量〕)和UV-XEFL(主波长峰为320nm、4.5W〔使用1.5W/cm的长度3cm的量〕)。
超声波清洗机:
US-18KS(SND Co.,Ltd.制)
卡尔费休水分计:
MKC-510(京都电子工业株式会社制)
液相色谱仪(HPLC):
HPLC:Prominence(岛津制作所制)
实施例1-1:由吡啶和柠康酸形成的晶体(8)的制造(其一)
将柠康酸(3.11g,23.90mmol)和吡啶(1.89g,23.89mmol)依次添加至乳钵中,将生成的固体用碾槌碾磨5分钟并充分混合,从而得到白色固体(4.79g)形式的由吡啶和柠康酸形成的晶体(8)(收率为96%)。
针对由吡啶和柠康酸形成的晶体(8),进行下述分析。
固相13C-NMR:
4mm CP/MAS probe,13C,CP/TOSS法,接触时间4ms,转速8kHz,
外部基准试样adamantane(29.472ppm)
δ172.4,168.4,144.4,144.4,142.5,140.1,134.2,129.8,127.9,25.8ppm
FT-IR:
图1为晶体(8)的FT-IR谱图。
粉末X射线衍射:
<粉末X射线衍射的分析条件>
X射线:Cu-Kα
电压:40kV
电流:15mA
步宽:0.020deg
扫描范围:2θ=3~40deg
图2为晶体(8)的粉末X射线衍射谱图。由所述粉末X射线衍射谱图读取的粉末X射线衍射的峰值如下所示。
2θ=12.56,15.03,16.06,17.58,18.29,19.21,21.55,22.99,24.50,26.43,27.04,28.08,30.33,32.48,34.69,35.87,36.44,38.43
实施例1-2:晶体(8)的制造(其二)
将柠康酸(130.1mg,1.00mmol)、吡啶(80.6μL,1.00mmol)和甲醇(2mL)依次添加至螺口瓶(Maruemu Corporation制造的螺纹管)中,混合该反应混合物而使其溶解。接着,将该放有反应混合物的螺口瓶的口用纱布覆盖,在运转中的气流室内以20℃~25℃静置64小时,使甲醇蒸发,从而得到白色固体(197.2mg)形式的由吡啶和柠康酸形成的晶体(8)(收率为94%)。
针对由吡啶和柠康酸形成的晶体(8),进行下述分析。
粉末X射线衍射:
<粉末X射线衍射的分析条件>与实施例1-1记载的条件相同。
以下示出晶体(8)的粉末X射线衍射的峰值。
2θ=12.56,15.04,16.08,17.58,18.33,19.14,21.55,23.01,24.43,26.42,27.03,28.07,30.40,32.48,34.70,35.88,36.44,38.42
单晶X射线结构分析:
使用Bruker公司制造的单晶X射线衍射计SMART APEX II ULTRA,利用Cu-Kα射线(波长:),冷却至-50℃并测定。X射线衍射数据的积分处理使用SAINT软件,空间群确定和晶体结构分析使用SHELXTL-97程序,进行上述作为白色固体的晶体(8)的单晶X射线结构分析。表5示出晶体(8)的晶体数据和结构精密化。
<单晶X射线结构分析的分析条件>
X射线:Cu-Kα
电压:50kV
电流:24mA
测定温度:-50℃
<晶体(8)的晶体数据和结构精密化>
〔表5〕
基于晶体(8)的单晶X射线结构分析结果,图3利用圆柱模型一并示出柠康酸的阴离子与吡啶嗡的堆积结构以及晶胞。
图3中,碳原子为黑色、氢原子为白色、氮原子和氧原子在图中用元素符号来表示,图中的2条点划线表示最接近的双键彼此。
进而,基于晶体(8)的单晶X射线结构分析结果,图4以ORTEP图的形式仅示出晶体(8)中的柠康酸的阴离子彼此的构型。
根据上述单晶X射线结构分析的结果,晶体(8)是仅由摩尔比为1:1的柠康酸的阴离子与吡啶嗡这两种构成要素组成的晶体。
进而,根据图4的结果,在晶体(8)的晶体结构中,最接近的双键彼此平行地存在,用下述式(8A)表示的平行双键间的距离L1为
实施例1-3:晶体(8)的制造(其三)
向反应容器中添加庚烷(28mL)和吡啶(1.87mL,23.25mmol),将所得溶液保持在20℃~25℃。接着,耗费10分钟向该溶液中滴加溶解于醋酸乙酯(28mL)的柠康酸(2.75g,21.14mmol),以20℃~25℃搅拌20分钟,制备由吡啶和柠康酸形成的晶体(8)的浆料。停止搅拌后,将过滤出的固体用庚烷(15mL)与醋酸乙酯(15mL)的混合溶剂进行清洗,接着,用己烷(30mL)清洗,并进行真空干燥。由此,以白色固体(4.14g)的形式得到由吡啶和柠康酸形成的晶体(8)(收率为94%)。
针对由吡啶和柠康酸形成的晶体(8),进行下述分析。
粉末X射线衍射:
<粉末X射线衍射的分析条件>与实施例1-1记载的条件相同。
以下示出晶体(8)的粉末X射线衍射的峰值。
2θ=12.61,15.08,16.11,17.64,18.39,19.26,21.61,23.05,24.57,26.51,27.12,28.15,30.43,32.53,34.74,35.94,36.50,38.44
根据上述粉末X射线衍射的结果,通过实施例1-1~1-3得到的晶体可视为相同的物质。此外,上述式(8)表示由吡啶和柠康酸形成时的吡啶/柠康酸(1:1)的晶体。
实施例2-1:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其一)
将由吡啶和柠康酸形成的晶体(8)(100.0mg、0.478mmol)铺展在玻璃制培养皿上,盖上培养皿的盖子。将该培养皿置于设定为25℃的冷板(cool plate)上,用100W的高压汞灯进行50小时的光照射来进行环化反应。培养皿与光源的距离设为1cm。停止反应后,以淡黄色固体(80.2mg)的形式回收反应混合物。根据该反应混合物的分析,目标化合物(13a)的收率为83%、转化率为94%、选择性>99%。收率通过使用马来酸作为标准品的定量1H-NMR来确定。另外,转化率和选择性根据GC分析中的目标化合物、无用的非对映体、源自原料的峰的相对面积比来算出。
用于求出上述环化反应中的转化率和选择性的GC样品的制备的相关概要示于(式A)。
为了求出转化率和选择性,在使用晶体(8)的光环化反应后,采取反应混合物的一部分,进行后述GC样品制备法B,由目标化合物(13a)及其非对映体衍生成化合物(14a)及其非对映体后,进行GC分析和GC-HRMS分析。
接着,用GC分析来追踪与目标化合物(13a)的非对映体相当的副产物,因此,通过与使用晶体(8)的光环化反应不同的方法,进行以柠康酸酐(11)作为起始物质的溶液中的光环化反应来作为工序(z),从而合成化合物(12a)及其非对映体。进行后述的GC样品制备法A,针对化合物(12a)及其非对映体进行水解反应、接着进行甲酯化反应,从而经由目标化合物(13a)及其非对映体而衍生成化合物(14a)及其非对映体,然后进行GC分析和GC-HRMS分析。应予说明,柠康酸酐溶液中的照射光的环化反应的有关详情在后述的参考例中进行叙述。
记载GC分析条件和GC-HRMS分析条件。
<GC分析条件>
柱:TC-5(0.53mm×30m、膜厚1.5μm)
载气:氦气
流量:3.3mL/min(恒定流量)
狭缝比:1/10、试样注入量:3μL
柱温:80℃(保持2分钟),升温速度:10℃/分钟、250℃(保持11分钟)
注入口温度:280℃
检测器温度:280℃
<GC-HRMS分析条件>
柱:DB-5(0.25mm×30m、膜厚0.25μm)
载气:氦气
流量:1mL/min(恒定流量)
狭缝比:1/50、试样注入量:0.2μL
柱温:80℃(保持3分钟),升温速度:25℃/分钟、250℃(保持7.2分钟)
注入口温度:280℃
离子化方法:EI、CI+
记载GC样品制备法和分析结果。
<GC样品制备法A>
该制备法是(式A)记载的制备工序I、后续的制备工序II。照射光的环化反应后,将蒸馏去除了溶剂的反应混合物(20mg)采取至螺口瓶中,添加甲醇(3mL)和0.05mol/L的氢氧化钠水溶液(2mL)后,将该溶液以20℃~25℃搅拌20分钟。将该反应混合物(1mL)的一部分取出并置于螺纹管中,添加甲苯(0.2mL)后,添加己烷溶液的三甲基甲硅烷基重氮甲烷(0.2mL、约0.6mol/L、东京化成工业株式会社制造的市售品),以20℃~25℃搅拌20分钟。将该反应混合物的有机层(0.4mL)取出一部分,用甲醇(1mL)稀释,将如此得到的溶液作为GC分析样品。
如(式A)所示那样,化合物(12a)通过GC样品制备法A而衍生成化合物(14a)。记载其分析结果。
GC分析:保留时间=18.62分钟
GC-HRMS分析:m/z calcd for C16H25O8[M+C2H5]+:345.1549,found345.1577
化合物(12a)的非对映体是化合物(12b)、化合物(12c)或化合物(12d)之一,利用GC样品制备法A衍生成化合物(14b)、化合物(14c)或化合物(14d)之一。记载其分析结果。
GC分析:保留时间=18.97分钟
GC-HRMS分析:m/z calcd for C16H25O8[M+C2H5]+:345.1549,found345.1561
根据由GC-HRMS测得的质量数结果可以确认到:与化合物(14a)的分子量相同但GC的保留时间不同的该非对映体。
<GC样品制备法B>
该制备法是(式A)记载的制备工序II。进行照射光的环化反应后,采取经过滤操作等而取出的反应混合物(25mg),添加甲醇(0.5mL)和甲苯(0.5mL)来制备溶液。接着,向该溶液中添加己烷溶液的三甲基甲硅烷基重氮甲烷(0.8mL、约0.6mol/L、东京化成工业株式会社制造的市售品),以20℃~25℃搅拌20分钟。将该反应混合物(0.12mL)取出一部分,用甲醇(1.5mL)稀释,将如此得到的溶液作为GC分析样品。
化合物(13a)通过GC样品制备法B而衍生成化合物(14a)。记载其分析结果。
GC分析:保留时间=18.62分钟
GC-HRMS分析:m/z calcd for C16H25O8[M+C2H5]+:345.1549,found345.1542
未反应的柠康酸通过GC样品制备法B而衍生成(Z)-2-甲基-2-丁烯二酸二甲酯。记载其分析结果。
GC分析:保留时间=9.68分钟
GC-HRMS分析:m/z calcd for C9H15O4[M+C2H5]+:187.0970,found187.0972
转化率以基于GC分析结果的(Z)-2-甲基-2-丁烯二酸二甲酯、化合物(14a)及其非对映体的相对面积比来算出。
选择性以基于GC分析结果的化合物(14a)及其非对映体的相对面积比来算出。
实施例2-2:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其二)
将由吡啶和柠康酸形成的晶体(8)(5.00g、23.90mmol)以及作为溶剂的己烷(100mL)添加至能够用磁力搅拌器搅拌的光化学反应实验装置(SEN LIGHTS Co.,Ltd.制)中,将光源设置于内部的中央。将反应温度设定为20℃~25℃并搅拌浆料,用100W的高压汞灯进行20小时的照射光的环化反应。停止反应后进行过滤,从而回收白色固体的反应混合物(4.33g)。根据该反应混合物的分析,目标化合物(13a)的收率为94%、转化率为95%、选择性>99%。收率通过使用马来酸作为标准品的定量1H-NMR来确定。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比算出。
实施例2-3:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其三)
将由吡啶和柠康酸形成的晶体(8)(25.00g、119.50mmol)、庚烷(200mL)和醋酸正丁酯(200mL)添加至能够用磁力搅拌器搅拌的光化学反应实验装置(SEN LIGHTS Co.,Ltd.制)中,将光源设置于内部的中央。将反应温度设定为20℃~25℃并搅拌浆料,用400W的高压汞灯进行12小时的照射光的环化反应。停止反应后进行过滤,从而回收白色固体的反应混合物(19.64g)。根据该反应混合物的分析,目标化合物(13a)的收率为93%、转化率为95%、选择性>99%。收率通过该反应混合物的收量和1H-NMR来算出。转化率和选择性根据实施例2-1记载的方法来分析,由GC分析的各种峰的相对面积比来算出。
对上述光反应后得到的白色固体的反应混合物进行分离精制。关于精制,从该反应混合物取出10.00g,溶解至甲醇(70mL)中,进行过滤后,蒸馏去除溶剂。接着,溶解至醋酸(45mL)后,将该溶液以60℃搅拌2小时30分钟,然后冷却至20℃~25℃,并进行过滤。其后,蒸馏去除所得固体中包含的溶剂,使用真空泵进行真空干燥,从而得到白色固体形式的目标化合物(6.12g)。通过1H-NMR而算出的收率为83%。
采取该所得目标化合物的一部分,制备饱和溶解在加热至70℃的乙腈中的溶液。接着,将该溶液冷却至20℃~25℃,进行静置而重结晶,进行所得单晶的单晶X射线结构分析。
化合物(13a)的分析
1H NMR(DMSO-d6):
δ12.46(br),3.30(s,2H),1.42(s,6H)ppm
13C-NMR(DMSO-d6):
δ175.9,175.9,171.5,171.5,51.7,51.7,44.2,44.2,20.7,20.7ppm
单晶X射线结构分析:
基于化合物(13a)的单晶X射线结构分析结果,图5以ORTEP图的形式示出化合物(13a)的分子结构。
实施例2-4:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其四)
向能够用磁力搅拌器搅拌的光化学反应实验装置(SEN LIGHTS Co.,Ltd.制)中添加醋酸乙酯(110mL)和吡啶(7.49mL,93.01mmol),冷却至5℃。接着,耗费30分钟向该反应混合物中滴加溶解于醋酸乙酯(110mL)的柠康酸(11.00g,84.55mmol)。其后,将该反应混合物以5℃搅拌20分钟,制备由吡啶和柠康酸形成的晶体(8)的浆料。向该反应容器中插入100W的高压汞灯,以5℃一边搅拌一边进行22小时的照射光的环化反应。其后,利用桐山漏斗滤取固体,用醋酸乙酯(30mL)清洗并真空干燥,从而得到白色固体的反应混合物(12.82g)。根据该反应混合物的分析,目标化合物(13a)的收率为93%、转化率为95%、选择性>99%。收率通过该反应混合物的收量和1H-NMR来算出。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比算出。
实施例3-1:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造
将由吡啶和柠康酸形成的晶体(8)(200.0mg、0.956mmol)以及作为溶剂的醋酸乙酯(4mL)添加至螺口瓶(Maruemu Corporation制造的螺纹管)中,从而制备浆料。其后,以能够用磁力搅拌器进行搅拌的方式进行操作,将光源设置于螺口瓶的外部。螺口瓶与光源的距离设为4.5cm。用100W的高压汞灯以20℃~25℃进行20小时的照射光的环化反应。停止反应后进行过滤,得到白色固体的反应混合物(116.0mg)。根据该反应混合物的分析,目标化合物(13a)的收率为69%、转化率为97%、选择性>99%。收率通过该反应混合物的收量和1H-NMR来算出。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比来算出。
除了将溶剂醋酸乙酯变更成表6记载的溶剂之外,利用与实施例3-1记载的反应相同的条件来实施反应。将所用的溶剂、转化率、选择性、收率和回收时的外观的有关实验结果作为实施例3-2~3-8来示于表6。
表6中,AcOn-Bu表示醋酸正丁酯,hexane/AcOn-Bu(v/v=1/1)表示己烷(2mL)与醋酸正丁酯(2mL)的混合溶剂,heptane/AcOn-Bu(v/v=1/1)表示庚烷(2mL)与醋酸正丁酯(2mL)的混合溶剂。
〔表6〕
实施例4:(3aR,3bR,6aS,6bS)-3a,6a-二甲基环丁烷[1,2-c:3,4-c’]二呋喃-1,3,4,6(3aH,3bH,6aH,6bH)-四酮(12a)的制造
[工序(a)和工序(b)的连续化]
作为工序(a),将吡啶(60mL)和柠康酸(15.55g、119.52mmol)依次添加至能够用磁力搅拌器搅拌的光化学反应实验装置(SEN LIGHTS Co.,Ltd.制)中,以20℃~25℃搅拌10分钟。搅拌结束后,添加作为溶剂的庚烷(175mL)和醋酸正丁酯(175mL),搅拌30分钟,制备由吡啶和柠康酸形成的晶体(8)的浆料。接着,将光源设置于该反应容器的内部中央,作为工序(b),将通过工序(a)得到的晶体(8)的浆料以20℃~25℃用400W的高压汞灯进行8小时的光照射并搅拌。通过停止光照射来终止反应后,对所得浆料进行过滤,从而得到白色固体的反应混合物。根据该反应混合物的分析,目标化合物(13a)的转化率为97%、选择性>99%。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比来算出。
为了对光反应后得到的粗产化合物(13a)进行精制,使该反应混合物溶解于四氢呋喃(250mL)并过滤后,蒸馏去除溶剂。接着,使用醋酸(40mL)和醋酸正丁酯(40mL)的混合溶剂进行悬浮清洗,再次进行过滤。其后,使用真空泵进行真空干燥,从而得到精制的化合物(13a)。
[工序(c)]
将上述精制后的化合物(13a)、甲苯(60mL)和醋酸酐(31.8mL、336.4mmol)添加至反应容器中,以100℃搅拌3小时。搅拌结束后,通过将反应容器冷却至20℃~25℃而终止反应。对所得反应混合物进行过滤后,使用醚溶剂来清洗所得固体,使用真空泵进行真空干燥,从而得到白色固体形式的目标化合物(12a)(11.36g)。通过1H-NMR算出的源自起始原料柠康酸的总收率为85%。采取该所得目标化合物的一部分,进行单晶X射线结构分析。
化合物(12a)的分析结果如下所示。
1H NMR(DMSO-d6):
δ3.88(s,2H),1.38(s,6H)ppm
13C-NMR(DMSO-d6):
δ173.5,173.5,168.1,168.1,49.0,49.0,44.1,44.1,15.7,15.7ppm
单晶X射线结构分析:
基于化合物(12a)的单晶X射线结构分析结果,图6以ORTEP图的形式示出化合物(12a)的分子结构。
实施例5:使用了光敏剂的(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造
将作为光敏剂的二苯甲酮(21.78mg、0.12mmol)和作为溶剂的庚烷(10mL)添加至螺口瓶(Maruemu Corporation制造的螺纹管)中。确认二苯甲酮溶解后,向螺口瓶中添加由吡啶和柠康酸形成的晶体(8)(500.0mg、2.39mmol)而制备浆料。制备结束后,以能够用磁力搅拌器搅拌的方式进行操作,将光源设置于螺口瓶的外部。螺口瓶与光源的距离设为4.5cm。用100W的高压汞灯以20℃~25℃进行5小时30分钟的照射光的环化反应。通过停止光照射来终止反应后,过滤所得浆料,从而得到白色固体的反应混合物(471.9mg)。根据该反应混合物的分析,目标化合物(13a)的转化率为34%、选择性>99%。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比来算出。
实施例6:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造
工序(a)
将中康酸(6M)(130.1mg、1.00mmol)、甲醇(2mL)和烟酰胺(9)(122.1mg、1.00mmol)依次添加至螺口瓶(Maruemu Corporation制造的螺纹管)中,混合该反应混合物而使其溶解。接着,将该放有反应混合物的螺口瓶的口用纱布覆盖,在运转中的气流室内以20℃~25℃静置64小时,使甲醇蒸发,从而得到白色固体形式的由烟酰胺和中康酸形成的晶体(10)。上述式(10)表示由烟酰胺和中康酸形成的烟酰胺/中康酸的晶体。
工序(b)
将由烟酰胺和中康酸形成的晶体(10)(100.0mg、0.396mmol)铺展在玻璃制培养皿上。盖上培养皿的盖子,并置于设定为25℃的冷板上,将培养皿与光源的距离设定为3cm后,用100W的高压汞灯进行20小时的光照射。通过停止光照射而终止反应后,回收白色固体形式的反应混合物。根据该反应混合物的分析,目标化合物(13a)的转化率为23%、选择性>99%。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比来算出。原料使用中康酸,GC样品制备中,未反应的中康酸衍生成(E)-2-甲基-2-丁烯二酸二甲酯。因此,(E)-2-甲基-2-丁烯二酸二甲至的峰视作源自原料的峰。
实施例7-1:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其一)
[工序(a)和工序(b)的连续化]
作为搅拌装置,使用带磁力搅拌器的光化学反应实验装置(SEN LIGHTS Co.,Ltd.制)。向该装置的反应容器中依次添加碳酸二甲酯(110mL)和吡啶(7.49mL、93.01mmol),冷却至10℃。冷却结束后,耗费30分钟向该反应溶液中滴加溶解于碳酸二甲酯(110mL)的柠康酸(11.00g、84.55mmol)。滴加结束后,将该反应混合物以10℃搅拌20分钟。搅拌结束后,得到由吡啶和柠康酸形成的晶体(8)的浆料。所得晶体(8)不进行分离/精制地直接用于后续工序。将该浆料以10℃进行搅拌,用100W的高压汞灯进行17小时的光照射。反应结束后,用漏斗滤取反应混合物中的固体后,用碳酸二甲酯(30mL)进行清洗。通过对所得固体进行真空干燥,从而得到白色固体形式的目标物14.23g。根据该白色固体的分析,目标化合物(13a)的收率为88%、转化率为94%、选择性>99%。收率通过该反应混合物的收量和1H-NMR分析来算出。转化率和选择性按照实施例2-1记载的方法,由GC分析的各种峰的相对面积比来算出。
实施例7-2:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其二)
[工序(a)和工序(b)的连续化]
作为搅拌装置,使用带磁力搅拌器的光化学反应实验装置(SEN LIGHTS Co.,Ltd.制)。向该装置的反应容器中添加醋酸乙酯(110mL)和吡啶(14.98mL、186.02mmol),冷却至5℃。冷却结束后,耗费30分钟向该反应溶液中滴加溶解于醋酸乙酯(110mL)的柠康酸(22.00g、169.10mmol)。滴加结束后,将该反应混合物以5℃搅拌20分钟。搅拌结束后,得到由吡啶和柠康酸形成的晶体(8)的浆料。所得晶体(8)不进行分离/精制地直接用于后续工序。将该浆料以5℃进行搅拌,用100W的高压汞灯进行33小时的光照射。光照射后的反应混合物中的水分量利用卡尔费休水分计(MKC-510、京都电子工业株式会社制)进行测定,结果为1570ppm。反应结束后,用漏斗滤取反应混合物中的固体后,用醋酸乙酯(40mL)进行清洗。通过将所得固体进行真空干燥,从而得到白色固体形式的目标物26.03g。根据该白色固体的分析,目标化合物(13a)的收率为93%、转化率为94%、选择性>99%。收率根据该反应混合物的收量和1H-NMR分析来算出。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比来算出。
实施例8-1:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其一)
将由吡啶和柠康酸形成的晶体(8)(100.0mg、0.478mmol)和醋酸乙酯(2mL)添加至螺口瓶(Maruemu Corporation制造的螺纹管)中,制备浆料。制备结束后,将该放有浆料的螺口瓶以其与光源的距离达到5cm的方式进行设置。设置结束后,作为光源而使用氙气灯(主波长峰为290nm、4.5W),进行2小时的光照射,使得浆料的温度保持在20℃~25℃。应予说明,搅拌使用磁力搅拌器。根据搅拌结束后的该浆料的HPLC分析,目标化合物(13a)的转化率为3%。
<HPLC分析条件>
检测器:差示折射率检测器
柱:Develosil C30-UG5(内径4.6mm、长度150mm、粒径5μm)
洗脱液:重量浓度为0.2%的三氟醋酸水溶液:乙腈=95:5(体积比)
流速:1.5mL/分钟、
柱温:35℃
保留时间:3.06分钟〔目标产物(13a)〕、3.39分钟〔柠康酸(6C)〕
实施例8-2:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造(其二)
将由吡啶和柠康酸形成的晶体(8)(300.0mg、1.434mmol)和醋酸乙酯(6mL)添加至螺口瓶(Maruemu Corporation制造的螺纹管)中,制备浆料。制备结束后,将该放有浆料的螺口瓶以其与光源的距离达到5cm的方式进行设置。设置结束后,作为光源而使用氙气灯(主波长峰为320nm、4.5W),进行77小时的光照射,使得浆料的温度保持在20℃~25℃。应予说明,搅拌使用磁力搅拌器。根据搅拌结束后的该浆料的HPLC分析(分析条件与实施例8-1相同),目标化合物(13a)的转化率为74%。
实施例9:(1R,2R,3S,4S)-1,3-二甲基环丁烷-1,2,3,4-四羧酸(13a)的制造
使用流动反应器来进行工序(a)和工序(b)。将流动反应器的概略图示于图7,将流动反应器中的具备双层管结构的T字型混合器(混合器2)的剖面图示于图8。流动反应器使用将内径2mm、外径3mm和长度10m的FEP管(由四氟乙烯/六氟丙烯共聚物形成的氟树脂制的管)缠绕于光源的灯夹套而得到的装置,将该装置设置于超声波清洗机。
工序(a):
制备浓度为0.34mol/L的柠康酸(437.9mg、3.366mmol)的醋酸乙酯溶液。此外,制备浓度为0.34mol/L的吡啶(266.3mg、3.366mmol)的醋酸乙酯溶液。将柠康酸的醋酸乙酯溶液和吡啶的醋酸乙酯溶液分别用注射泵以0.9mL/min的速度进行送液,在混合器1(图7中的21)中进行混合。其后,利用混合器2(图7中的32)来混合氮气,从而形成由晶体(8)的浆料和氮气得到的段塞流(浆料与氮气交替排列的流体)。应予说明,混合器2为了避免管的闭塞而具有图8所示的双层管结构,并设置在50℃的恒温槽中。
工序(b):
在基于450W的高压汞灯的光照射下,进一步在超声波照射下进行环化反应。利用质量流量控制器调整段塞流,使得其在11分钟内通过光和超声波的照射部分,来进行反应。根据所回收的流出液的HPLC分析,目标化合物(13a)的转化率为34%。
参考例1
按照前述非专利文献2,进行柠康酸酐(11)溶液中的照射光的环化反应。
将柠康酸酐(11)(1.38g、12.31mmol)、作为溶剂的1,4-二噁烷(10mL)和作为光敏剂的二苯甲酮(93.0mg、0.51mmol)添加至螺口瓶(Maruemu Corporation制造的螺纹管),以能够用磁力搅拌器进行搅拌的方式进行操作,将光源设置在螺口瓶的外部。将螺口瓶与光源的距离设为4.5cm。用100W的高压汞灯以20℃~25℃进行18小时的照射光的环化反应。停止反应后,根据反应混合物的分析,目标化合物(12a)的转化率为68%、选择率为50%。转化率和选择性由GC分析中的各种峰的相对面积比来算出。原料使用柠康酸酐,GC分析中,柠康酸酐的峰示作原料的峰。
GC分析和GC-HRMS分析用的样品的制备方法和分析结果如下所述。从悬浮的反应混合物中采取悬浮液(100μL),用二甲基亚砜(1.5mL)稀释,作为分析样品。
目标化合物(12a)的分析结果如下所示。
GC分析:保留时间=15.60分钟
GC-HRMS分析:m/z calcd for C10H9O6[M+H]+:225.0399,found 225.0386
目标化合物(12a)的非对映体的分析结果如下所示。
GC分析:保留时间=15.81分钟
GC-HRMS分析:m/z calcd for C10H9O6[M+H]+:225.0399,found 225.0403
根据由GC-HRMS测定的质量数结果可以确认到:与化合物(12a)的分子量相同但GC的保留时间不同的该非对映体。然而,无法确定化合物(12a)的非对映体的立体结构。推测目标化合物(12a)的非对映体为化合物(12b)、化合物(12c)或化合物(12d)中的一者。
参考例2
按照前述专利文献2记载的方法,进行柠康酸酐(11)溶液中的照射光的环化反应。
将柠康酸酐(11)(1.42g、12.67mmol)和作为溶剂的醋酸乙酯(10mL)添加至螺口瓶(Maruemu Corporation制造的螺纹管)中,以能够用磁力搅拌器进行搅拌的方式进行操作,将光源设置于螺口瓶的外部。未添加光敏剂,将螺口瓶与光源的距离设为4.5cm。用100W的高压汞灯以20℃~25℃进行60小时的照射光的环化反应。停止反应后,蒸馏去除反应混合物的溶剂,使用真空泵进行真空干燥,从而得到白色固体的反应混合物(1.36g)。根据该反应混合物的分析,目标化合物(12a)的转化率为88%、选择率为41%。GC样品的制备法使用前述GC样品制备法A。转化率和选择性按照实施例2-1记载的方法进行分析,并由GC分析的各种峰的相对面积比来算出。
前述专利文献2和非专利文献2中,针对刚刚进行照射光的环化反应之后、即进行精制操作之前的目标化合物和无用的非对映体的选择性没有记载。根据上述参考例的结果可确认:相对于目标化合物1,3-二甲基环丁烷-1,2,3,4-四羧酸二酐,无用的非对映体生成了1倍~1.4倍左右。因而,以往的制造方法中,由于选择性如此低,因此需要复杂的精制操作,推测其在生产效率的方面会造成不良影响。
产业上的可利用性
通过本发明得到的满足环丁烷环上的两个取代基位于1位和3位且该取代基的相对构型为反式的1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐作为聚酰亚胺等各种工业用的原料、合成中间体而在广泛的领域中得以应用,是有用的化合物。
应予说明,2014年5月9日申请的日本专利申请2014-098037号的说明书、权利要求书、附图和摘要的全部内容援引至此,作为本发明说明书的公开内容并入。
附图标记说明
11:放有化合物(6C)的醋酸乙酯溶液的注射泵
12:放有化合物(7)的醋酸乙酯溶液的注射泵
21:T字型混合器(混合器1)
31:恒温槽
32:具有双层管结构的T字型混合器(混合器2)
33:质量流量控制器
34:氮气瓶
41:超声波清洗机
42:灯夹套
43:光源
44:回收目标生成物的容器
- 还没有人留言评论。精彩留言会获得点赞!