一种基因定位整合表达系统及其应用的制作方法
本发明涉及生物技术领域,具体地涉及一种利用基因工程技术制备的基因定位整合表达系统及其应用。
背景技术:
转基因动物表达的外源蛋白具有精确的转录后加工,翻译后修饰(糖基化,二硫键形成等),表达产物在分子结构、理化性质各生物学功能等方面与天然蛋白更接近。在制备一些翻译后修饰较复杂的重组蛋白时,往往利用转基因哺乳动物表达系统产生的重组蛋白具有较好的活性。但是,与其它表达系统相比,实现重组蛋白在转基因动物中高表达存在研制周期长,综合成本高等问题,这主要是由于目前采用的随机转基因方法存在外源基因在基因组中的整合位点和拷贝数不可控、外源基因在动物体内表达不稳定,转基因动物个体之间表型差异等问题,严重制约了转基因动物表达系统的应用。
研制高效、经济的转基因动物表达系统一直是生物工程技术领域,尤其是转基因动物生物反应器领域的一个主要发展方向。研究表明,外源基因表达特性与其所处的基因位点密切相关,整合于常染色质区的外源基因一般能获得较好的表达,而当外源基因整合在异染色质附近时就会出现不表达或嵌合表达的现象。有数据显示拷贝数与表达水平之间没有明显联系,而整合位点则是影响表达的主要因素。在转基因鱼、转基因奶山羊和转基因奶牛中外源基因的整合与表达也因整合位点不同而存在差异。此外,转基因的随机整合还可能会引起插入突变,产生明显的表型效应。
外源基因整合位点对于转基因的表达至关重要,开发一种高效转基因动物表达系统的必要条件是找到一个合适的区域用于外源基因定位表达。在以往的研究中,人们发现有一些基因位点可支持外源基因高表达,如β-肌动蛋白(β-actin)位点、次黄嘌呤磷酸(hprt)位点等。但这些位点用于支持外源基因表达尚存在一些不足之处:β-肌动蛋白定点整合的转基因小鼠因β-肌动蛋白的缺失而产生了一些异常现象;次黄嘌呤磷酸(hprt)位点位点位于x染色体上,在雌性动物中常会由于随机x染色体失活而导致该位点的失活。
综上所述,本领域迫切需要开发适用于外源基因定位表达的整合位点,建立高效的定位整合表达系统。
技术实现要素:
本发明的目的在于提供建立一种高效的转基因羊定位整合表达系统,解决因转基因整合位点、整合拷贝数不确定带来的外源基因表达不确定问题,使转基因表达变得可控可期。
在本发明的第一方面,提供了一种核酸构建物,所述的构建物具有从5到3’的式i结构:
a5-b5-c-b3-a3-d(i)
式中,
a5、b5、c、b3、a3、d分别为用于构成所述构建物的元件;
各“-”独立地为键或核苷酸连接序列;
a5为5’同源臂序列;
b5为5’位点特异性重组序列;
c为串联的、各自独立的外源基因表达盒和第一筛选标记基因的表达盒;
b3为3’位点特异性重组序列;
a3为3’同源臂序列;
d为无或第二筛选标记基因的表达盒;
并且,所述的5’同源臂序列和3’同源臂序列使得所述构建物与山羊染色体发生定点重组,且所述的定点重组的位点位于山羊5号染色体mical3和pex26之间的区域(nc_022297.1第100383471-100500925位)”。
在另一优选例中,所述的5’同源臂序列和3’同源臂序列分别结合于山羊5号染色体mical3和pex26之间、且位于所述定点重组的位点(即重组位点)两侧的区域。
在另一优选例中,所述的5’同源臂序列和/或3’同源臂序列的长度为200-5000bp,较佳地为500-3000bp,更佳地为1000-2000bp。
在另一优选例中,所述的5’同源臂序列选自:
(a1)seqidno:1所示的核苷酸序列;或
(a2)与(a1)中的核苷酸序列互补的核苷酸序列。
在另一优选例中,所述的3’同源臂序列选自:
(b1)seqidno:2所示的核苷酸序列;或
(b2)与(b1)中的核苷酸序列互补的核苷酸序列。
在另一优选例中,所述的5’同源臂序列如下制备:
以seqidno:11和seqidno:12为引物,以山羊基因组dna为模板pcr扩增获得。
在另一优选例中,所述的3’同源臂序列如下制备:
以seqidno:13和seqidno:14为引物,以山羊基因组dna为模板pcr扩增获得。
在另一优选例中,所述的位点特异性重组序列选自下组:loxp、和flp。
在另一优选例中,所述的5’位点特异性重组序列选自:
(c1)seqidno:3所示的核苷酸序列;或
(c2)与(c1)中的核苷酸序列互补的核苷酸序列。
在另一优选例中,所述的3’位点特异性重组序列选自:
(d1)seqidno:4所示的核苷酸序列;或
(d2)与(d1)中的核苷酸序列互补的核苷酸序列。
在另一优选例中,所述的外源基因选自下组:红色荧光蛋白基因、溶菌酶基因、人乳清白蛋白基因、全人源肿瘤细胞坏死因子单克隆抗体基因、绿色荧光蛋白基因、和血清白蛋白基因。
在另一优选例中,所述的筛选标记选自下组:嘌呤霉素抗性基因、杀稻瘟菌素s脱氨酶基因(bsr)、和新霉素基因。
在另一优选例中,所述的核苷酸连接序列的长度为1-100bp,较佳地为1-50bp;更佳地为1-20bp。
在本发明的第二方面,提供了一种载体,所述的载体含有本发明第一方面所述的构建物。
在本发明的第三方面,提供了一种宿主细胞,所述的宿主细胞含有本发明第一方面所述的构建物,或其基因组整合有一个或多个本发明第一方面所述的构建物。
在另一优选例中,所述的宿主细胞为人或非人哺乳动物的细胞。
在另一优选例中,所述的非人哺乳动物选自:山羊、绵羊、猪、牛、狗和兔,较佳地为山羊。
在另一优选例中,所述的宿主细胞包括山羊成体体细胞、山羊胎儿体细胞、或山羊胚胎干细胞。
在另一优选例中,所述的宿主细胞包括山羊耳成纤维细胞。
在另一优选例中,所述的宿主细胞是用选自下组的方法将本发明第一方面所述的构建物导入细胞的:同源重组法、显微注射法、电穿孔法、脂质体转染法、磷酸钙沉淀法、病毒感染法、或精子载体法。
在本发明的第四方面,提供了一种体外制备转基因细胞的方法,包括步骤:
(i)将本发明第一方面所述的构建物、或含有所述构建物的载体转染细胞,使得所述构建物与所述细胞中的染色体发生定点重组,从而制得转基因细胞,且所述的定点重组的位点位于山羊5号染色体mical3和pex26之间的区域。
在另一优选例中,在步骤(i)中,还包括用进行定点切割的构建物对所述细胞进行转染,从而对所述细胞的染色体进行定点切割,其中定点切割的位点位于山羊5号染色体mical3和pex26之间的区域。
在另一优选例中,所述定点切割的位点位于本发明第一方面所述定点重组的位点附近。
在另一优选例中,所述的切割采用talen切割方法。
在另一优选例中,所述的talen切割方法采用ptl和ptr质粒。
在另一优选例中,所述的宿主细胞为人或非人哺乳动物的细胞。
在本发明的第五方面,提供了一种制备转基因动物的方法,包括步骤:
(i)将本发明第一方面所述的构建物、或含有所述构建物的载体转染细胞,使得所述构建物与所述细胞中的染色体发生定点重组,从而制得转基因细胞,且所述的定点重组的位点为山羊5号染色体mical3和pex26之间的区域;和
(ii)将获得的转基因细胞再生为动物体,从而获得转基因动物。
在另一优选例中,步骤(ii)包括步骤:
(ii-1)将所获得的转基因细胞作为核供体进行体细胞克隆,从而获得转基因动物。
在本发明的第六方面,提供了一种体外制备转基因细胞的方法,包括步骤:
(i)在cre重组酶存在的条件下,用一外源基因表达载体转化本发明第三方面所述的细胞,从而制得转基因细胞。
在另一优选例中,所述的外源基因表达载体具有从5到3’的式ii结构:
g1-h-g2(ii)
式中,
g1为5’位点特异性重组序列;
h为串联的、各自独立的外源基因表达盒和第一筛选标记基因的表达盒;
g2为3’位点特异性重组序列;
并且,所述的特异性重组序列与本发明第三方面所述细胞的基因组中的特异性重组序列匹配。
在另一优选例中,步骤(i)包括步骤:
(i-1)cre酶表达载体与外源基因表达载体共转化本发明第三方面所述的细胞;或
(i-2)在具有细胞穿透活性的tat-cre重组蛋白的作用下,用外源基因表达载体对本发明第三方面所述的细胞的染色体进行基因整合。
在本发明的第七方面,提供了一种制备转基因动物的方法,包括步骤:
(i)在cre重组酶存在的条件下,用一外源基因表达载体转化本发明第三方面所述的细胞,从而制得转基因细胞;和
(ii)将转化的细胞再生为动物体,从而获得转基因动物。
在本发明的第八方面,提供了一种转基因非人哺乳动物,所述的动物是用本发明第四方面或本发明第五方面所述的方法制备的。
在本发明的第九方面,提供了一种基因定点整合表达系统,通过转基因羊基因组中(nc_022297.1第100383471-100500925位)之间定点整合有本发明第一方面所述的构建物,以通过位点特异性重组实现其它外源的定位整合及有效表达。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
图1显示了靶向g6位点的含盒式交换loxp元件及dsred表达框的同源重组载体ptg6-dsred。
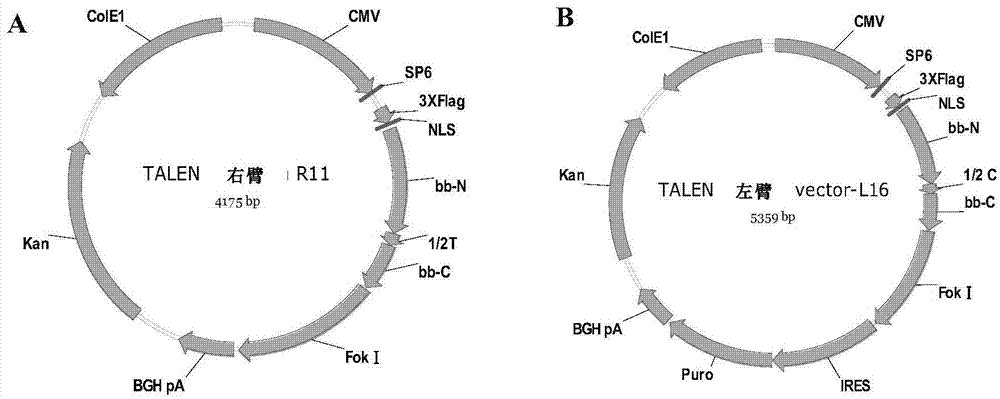
图2显示了talen左右臂表达载体的结构图。其中,图2a显示了talen右臂表达载体的结构图,图2b显示了talen左臂表达载体的结构图。
图3显示了各组合的talen表达载体在细胞中的活性。
图4显示了talen左右侧表达载体ptl、ptr的结构图。其中,图4a显示了talen左侧表达载体ptl的结构图,图4b显示了talen右侧表达载体ptr的结构图。
图5显示了重组载体ptg6-dsred在g6位点的同源重组示意图。
图6显示了g6位点发生同源重组的细胞基因型检测结果图。其中,图6a显示了打靶载体与基因组重组后,5'端用引物f6、r6进行pcr扩增的产物,图6b显示了3'端用引物f7、r7进行pcr的扩增产物。
图7显示了g6位点定位插入盒式交换元件loxp及dsred表达框的细胞克隆(g6-red-loxpc)。其中,图7a显示了中靶细胞的白光镜检图,图7b显示了对应的中靶细胞的红色荧光镜检图。
图8显示了g6位点定位插入盒式交换元件loxp及dsred表达框的转基因克隆羊(g6-red-loxpg)。
图9显示了g6-red-loxpg转基因克隆羊基因型分析结果图,其中,m为marker,,tg1为1号克隆羊,tg2为2号克隆羊,n为野生型羊。图9a显示了用5'同源臂外引物(g6-5f)与目的基因启动子上引物(cmv-r)进行pcr的扩增产物,图9b显示了用嘌呤霉素上引物(puro-f399)与3'同源臂外引物的pcr(g6-3r)的扩增结果。
图10显示了分离自g6-red-loxpg转基因羊耳组织块的细胞团荧光镜检图。其中,图10a和图10c显示了转基因羊耳组织细胞团的白光镜检图,图10b和图10d分别显示了转基因羊耳组织细胞团的荧光镜检图。
图11显示了g6-red-loxpg转基因克隆羊部分组织中红色荧光rt-pcr检测结果图,gapdh为内参。其中,图11a为g6-red-loxpg转基因羊dsred的rt-pcr结果,图11b为野生型羊对照的rt-pcr结果;图11c为g6-red-loxpg转基因羊内参gadph的rt-pcr结果,图11d为野生型羊内参的检测结果。
图12显示了g6-red-loxpg转基因克隆羊各组织器官红色荧光镜检图。
图13显示了含盒式交换元件loxp及gfp表达框的重组载体ptg6-gfp。
图14显示了cre介导转基因细胞中g6位点发生盒式基因交换示意图。
图15a和图15b显示了重组细胞克隆的基因型检测结果图。
图16显示了g6位点定位重组loxp及gfp表达框的细胞克隆(g6-gfp-loxpc)。其中,图16a显示了重组细胞克隆的白光镜检图,图16b显示了对应重组细胞的荧光镜检图。
具体实施方式
本发明人经过广泛而深入地研究,首次意外地发现在羊基因组中的5号染色体上mical3及pex26基因之间存在一个染色体区域(命名为g6位点,ncbireferencesequence:nc_022297.1),所述的染色体区域可支持外源基因的高效稳定表达。
具体地,本发明人构建了一种核酸构建物,所述核酸构建物包含与所述染色体区域结合的同源臂序列,以及位点特异性重组序列、外源基因表达盒和筛选标记基因表达盒。将所述构建物导入宿主细胞,可以实现目的基因在宿主细胞基因组中的定位整合与表达,获得转基因细胞。利用位点特异性重组技术,可以在所述转基因细胞中,实现其它特定外源基因的表达及转基因的遗传修饰。
术语
如本文所用,术语“同源臂”指打靶载体上待插入的外源序列两侧的与基因组序列完全一致的侧翼序列,用于识别并发生重组的区域。
如本文所用,术语“位点特异性重组序列”指一种特殊的遗传重组系统,区别于常规的遗传重组,位点特异性重组主要依赖于特定的位点特异性重组酶(site-specificrecombinases,ssrs),ssrs通过识别并结合到特定的序列,使序列间的dna发生特定重排。
如本文所用,术语“loxp元件”指可以被cre重组蛋白识别的两个同向重复位点。
如本文所用,术语“cre酶”指介导2个loxp位点之间特异性重组的蛋白酶,它可以导致loxp位点之间的核苷酸序列被删除或重组。
如本文所用,“外源基因”指作用是阶段性作用的外源dna分子。可用于本申请的外源基因没有特别限制,包括转基因动物领域常用的各种外源基因。代表性例子包括(但并不限于):红色荧光蛋白基因、绿色荧光蛋白基因、溶菌酶基因、鲑鱼降钙素基因、乳铁蛋白、或血清白蛋白基因等。
如本文所用,“筛选标记基因”指转基因过程中用来筛选转基因细胞或转基因动物的基因,可用于本申请的筛选标记基因没有特别限制,包括转基因领域常用的各种筛选标记基因,代表性例子包括(但并不限于):新霉素基因、或嘌呤霉素抗性基因。
如本文所用,术语“表达盒”是指含有待表达基因以及表达所需元件的序列组件的一段多聚核苷酸序列。例如,在本发明中,术语“筛选标记表达盒”指含有编码筛选标记的序列以及表达所需元件的序列组件的多聚核苷酸序列。表达所需的组件包括启动子和聚腺苷酸化信号序列。此外,筛选标记表达盒还可以含有或不含有其他序列,包括(但并不限于):增强子、分泌信号肽序列等。
在本发明中,适用于外源基因表达盒和筛选标记基因表达盒的启动子可以是任何一种常见的启动子,它可以是组成型启动子或诱导型启动子。较佳地,该启动子是组成型的强启动子,例如牛β-乳球蛋白启动子等其它适用于真核表达的启动子。
talen技术
tale(transcriptionactivator-likeeffector)是由植物致病细菌xanthomonas分泌的一类具有转录激活功能的蛋白,该种蛋白通过其内部保守的重复氨基酸序列与植物宿主基因启动子区的相应核苷酸序列发生特异性结合,并激活基因表达。tale的dna结合域由一些非常保守的重复氨基酸序列模块组成,每个模块一般包含33~35个氨基酸(aa),其第12,13位氨基酸种类可变且决定了tale与dna结合的特异性。因此称这种重复可变的双氨基酸序列为rvd(repeatvariablediresidue)。
talen(transcriptionactivator-likeeffectornuclease)是一种人工改造的限制性内切酶,是将tale的dna结合域与限制性内切酶(fokⅰ)的dna切割域融合而得到。tale的dna结合域中的重复氨基酸序列模块可与单碱基发生特异性结合,从而选择靶dna序列进行改造,是一种非常有效的基因组改造工具酶。
cre-loxp系统
cre重组酶基因编码区序列全长1029bp(embl数据库登录号x03453),编码38kda蛋白质。cre重组酶是一种由343个氨基酸组成的单体蛋白。属于λint酶超基因家族,它不仅具有催化活性,而且与限制酶相似,能识别特异的dna序列,即loxp位点,使loxp位点间的基因序列被删除或重组。
loxp(locusofx-overp1)序列来源于p1噬菌体,是有两个13bp反向重复序列和中间间隔的8bp序列共同组成,8bp的间隔序列同时也确定了loxp的方向。cre酶在催化dna链交换过程中与dna共价结合,13bp的反向重复序列是cre酶的结合域。
本发明所用的loxp序列包括野生型loxp位点序列和突变的loxp位点序列。
在一种优选的实施方式中,本发明所用的两个突变型loxp位点序列为lox2272及lox66,其中,lox2272序列为:ataacttcgtataggatactttatacgaagttat(seqidno.:3);lox66序列为:ataacttcgtatagcatacattatacgaacggta(seqidno.:4)。
核酸构建物
本发明还提供了一种构建物,所述的构建物如本发明第一方面所述。
本发明的构建物中所用的各种元件都是本领域中已知的,因此本领域技术人员可以用常规方法,如pcr方法、全人工化学合成法、酶切方法获得相应的元件,然后通过熟知的dna连接技术连接在一起,就形成了本发明的构建物。
将本发明的构建物插入外源载体(尤其是适合转基因动物操作的载体),就构成了本发明的载体。
将本发明的载体转化宿主细胞从而介导本发明的载体对宿主细胞染色体进行整合,制得转基因细胞。
本发明还提供了一种定位整合外源基因的方法,包括步骤:
(i)在cre重组酶存在的条件下,用一外源基因表达载体转化本发明第三方面所述的细胞,从而制得转基因细胞。
在另一优选例中,所述的外源基因表达载体包含特异性重组序列并且与本发明第三方面所述细胞基因组中的特异性重组序列匹配。
本发明的主要优点包括:
(a)本发明的基因定点整合区域可用于外源基因的高效表达。
(b)本发明第三方面细胞的基因组特定区域整合有位点特异性重组序列,可用于外源基因的定点整合和高效表达。
(c)本发明获得的定位表达红色荧光蛋白的转基因羊可作为一种工具羊,用于其它外源基因的定点整合与替换表达。
(d)本发明获得的转基因羊定位表达系统可作其它特定外源基因的高效表达。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
实施例1
含loxp序列的同源重组表达载体ptg6-dsred的构建
设计g6位点基因打靶载体,以红色荧光蛋白dsred为目的基因,以嘌呤霉素为正筛选基因,白喉毒素为负筛选基因构建质粒,在目的基因两侧连接两个含不同核心序列的loxp位点(lox2272,lox66),利用同源重组实现loxp序列在g6位点的锚定。
具体地,以pko-v913为骨架载体,在其多克隆位点以rsrii单酶切插入负筛选基因白喉毒素(diphtheriatoxina,dt),获得pkov913-dt。
以质粒pdsred-n1为模板,用含lox2272的引物f1:ccgctcgagataacttcgtataggatactttatacgaagttattagttattaatagtaatcaat(seqidno.:5),引物r1:gaatgatatcgatctgacggttcactaaaccag(seqidno.:6)进行pcr,扩增642bp的含cmv基因启动子片段1。
以质粒pdsred-n1为模板,用引物f2:gaatgatatcgccaccatggcctcctccgagaacg(seqidno.:7),引物r2:cgggatccgatgagtttggacaaaccac(seqidno.:8)进行pcr,扩增含红色荧光蛋白表达框dsred的基因片段2。将片段1用xhoi+ecorv酶切,片段2用ecorv+bamhi双酶切后,连入经xhoi+bamhi双酶切的pkov913-dt载体,获得pkov913-dt-dsred。
以改造后的质粒pkopurov810(删去了xhoi及hindiii酶切位点)为模板,用引物f3:cgggatccctaccgggtaggggaggcgctt(seqidno.:9),含lox66的引物r3:cccaagctttaccgttcgtataatgtatgctatacgaagttatgccccagctggttctttccgc(seqidno.:10)进行pcr,扩增1.5kb的抗性基因嘌呤霉素表达框片段3。用bamhi+hindiii酶切片段3,连入同样经bamhi+hindiii双酶切的载体pkov913-dt-dsred,获得含抗性基因载体pkov913-dt-dsred-puro。
以山羊胎儿成纤维细胞g114cs基因组dna为模板,用引物f4:ggggtaccctccctcagtatccaagctatcc(seqidno.:11),引物r4:ccgctcgagaaaggtgggaagggaactaatttat(seqidno.:12)进行pcr,扩增565bp的5′同源臂片段4。用kpni+xhoi酶切片段4,将其连入同样经kpni+xhoi双酶切的载体pkov913-dt-dsred-puro,获得含左侧同源臂载体pkov913-dt-l-dsred-puro。
以山羊胎儿成纤维细胞g114cs基因组dna为模板,用引物f5:cccaagctttcaacgcaagcaaggttttaagc(seqidno.:13),引物r5:acgcgtcgacttctctggtgctctgatgggt(seqidno.:14)进行pcr,扩增1097bp的3′同源臂片段5。用hindiii+sali酶切片段5,将其连入同样经hindiii+sali双酶切的载体pkov913-dt-l-dsred-puro,获得含双侧同源臂的打靶载体pkov913-dt-l-dsred-puro-r,最终命名为ptg6-dsred,质粒图谱如图1所示。
实施例2
talen左右臂表达载体的构建及活性验证
完全的talen序列包括左边的结合区域、切割区域、和右边的结合区域。参照g6位点基因序列,根据talen靶位点设计的原则,选择相隔17-18bp的两段14-19bp的序列作为靶位点,分别设计三条左臂及两条右臂,具体序列如下图如示:
l1:gctcctgataaattagttc(seqidno.:15);
l2:gctcctgataaattagtt(seqidno.:16);
l3:gctcctgataaattagt(seqidno.:17);
r1:tgcttgcgttgaggtat(seqidno.:18);
r2:gcttgcgttgaggtatt(seqidno.:19)。
分别对应上述靶点识别域,采用上海斯丹赛生物技术有限公司的talen骨架载体(图2)及基因克隆试剂盒进行基因克隆,构建了5个以cmv为启动子的talen真核表达载体。将克隆载体用bamhⅰ及pstⅰ双酶切后为两条带,分别为载体骨架及识别序列。将酶切正确的质粒送测序,测序结果与与设计的标准序列进行对比,最终获得正确的质粒ptl1,ptl2,ptl3,ptr1,ptr2。
为了验证talen表达质粒在山羊胎儿成纤维细胞中的活性,将构建的talen左右臂表达质粒组合后(ptl1-ptr1,ptl1-ptr2,ptl2-ptr1,ptl2-ptr2,ptl3-ptr1,ptl3-ptr2共六组)分别与prfp-puro质粒共转染山羊成纤维细胞,嘌呤霉素筛选5天后收集混合细胞克隆,抽提基因组dna后,pcr扩增靶位点附近序列,将扩增产物回收后加入t7e1酶切分析
结果见图3,6组talen组合均具活性,其中l1r2活性最高,l2r2活性最低;光密度扫描其活性为l1r2>l1r1/l3r1>l2r1/l3r2>l2r2。最终选择活性最好的ptl1-ptr2(图4)组合进行靶位点基因编辑。
实施例3
loxp锚定的表达红色荧光蛋白的细胞株制备
组织块法分离山羊胎儿成纤维细胞株,将其培养于含2mm的谷氨酰胺,1mm的丙酮酸钠,1×的非必需氨基酸,2ng/ml的碱性成纤维细胞生长因子,1000units/ml的小鼠白血病抑制因子,10%胎牛血清(gibco),100units/ml的青链霉素的glasgow基本培养基(gmem,gibco)中,细胞于37℃,5%co2培养箱中培养,传代二次后,取处于对数生长期的细胞准备进行基因转染。
图5显示了同源重组载体ptg6-dsred在g6位点的同源重组示意图。将构建的打靶载体ptg6-dsred经sali线性化,回收线性化载体dna片段15μg,将其与talen左右臂质粒(ptl1、ptr2)各7μg混合后共转染(lipo3000,invitrogen)第3代的山羊胎儿成纤维细胞。48小时后,将这些细胞消化后重新分到含0.8μg/mlpuromycin(sigma)的选择性培养液中。大约8-9天后,用克隆环将其中生长良好且独立的细胞克隆挑出,转移到48孔板孔。然后传代至24孔,待24孔板长满后,收集2/3细胞冻存,1/3用于抽提基因组dna。共挑取细胞克隆162株,其中148个长势良好,收集细胞,采用天根基因组dna提取试剂盒抽提细胞基因组dna备用。
分别在g6位点5′同源臂外、3′同源臂外,打靶载体内部设计两对引物检测中靶细胞株,位置见图5,引物序列如下:
f6:atgcgtgtatggagagagaaggt(seqidno.:20);
r6:tccccgcggtgcaggtcgaaaggcccggaga(seqidno.:21);
f7:tccccgcgggccaccatgaccgagtacaagcccacgg(seqidno.:22);
r7:atctctttgctgctgctctctga(seqidno.:23)。
以挑取的细胞克隆基因组dna为模板,分别用两对引物f6、r6及f7、r7扩增发生同源重组的细胞克隆的基因组dna片段,扩增条件:95℃5min;95℃30s,58℃30s,72℃,2/3min,30cycles;72℃,10min。理论上,阳性克隆用5′同源臂外引物f6、r6能扩增出的3.1kb条带,用3′同源臂外引物f7、r7能扩增出的2.2kb条带。
结果如下:
在148个细胞克隆中通过pcr检测获得17个中靶阳性克隆,同源重组效率为11.5%,选择1#克隆重复进行pcr检测,结果见图6;将pcr产物收集后进行序列测定,结果与预期相符,说明该细胞克隆在g6位点发生了同源重组,两个突变型loxp序列及红色荧光蛋白表达框已定点插入了目标染色体位点,该细胞命名为g6-red-loxpc。将该细胞克隆置于荧光显微镜下镜检,见细胞发出稳定的红色荧光(图7)。
实施例4
体细胞核移植制备loxp及红色荧光蛋白定位整合克隆羊及表达情况分析
用于提供核移植受体卵的羊、重构胚的临时培养寄母羊与受体羊均为萨能奶山羊。注射0.1mgpg使其情期同步,按常规应用fsh、gn-rh对供体羊超数排卵处理,回收体内成熟的卵母细胞,将其置于m16液中,在37.5℃,含5%的二氧化碳培养箱中作短暂培养待用。
准备同源重组的中靶细胞g6-red-loxpc,培养2-3d后丰度达80%时饥饿24-48hr;胰酶孵育吹吸细胞成单个细胞。卵母细胞经含细胞松弛素b的缓冲液洗后,将卵和供核细胞同时移入载玻片上的缓冲液中,在显微镜下去除卵母细胞的核,将供核体细胞移入卵周隙,培养后进行电融合与激活处理。
将激活后的重构卵包埋在0.8%琼脂糖中,移植到山羊的输卵管内继续培养。5天后,回收胚胎,观察琼脂糖块中核移植胚的发育情况。将其中发育至囊胚的重构胚2-3枚移植到发情后第六天的受体羊子宫内。试验共移植受体羊36头,获得怀孕羊20头,出生存活克隆羊2头(图8)。收集新生克隆羊血液5ml,采用天根基因组dna抽提试剂盒抽提基因组dna。分别采用两对引物g6-5f:atgcgtgtatggagagagaaggt(seqidno.:24),cmv-r:gatctgacggttcactaaaccag(seqidno.:25);puro-f399:caccagggcaagggtctgggca(seqidno.:26),g6-3r:atctctttgctgctgctctctga(seqidno.:27)对其进行pcr扩增,确定新生克隆羊基因型。结果见两头克隆羊均能扩增出相应的1.6kb及1.9kb的条带(图9),说明克隆羊基因组中g6位点定位整合有loxp序列及红色荧光表达框。
采用组织块法分离克隆山羊耳成纤细胞,将其置于37.5℃,含5%的二氧化碳培养箱中培养,数日后可见细胞游离出组织块,在荧光显微镜下观察时,细胞发出红色荧光(图10)。
分离出生后一周的转基因羊各组织器官,包括脑、心、肝、脾、肺、肾,采用rt-pcr分析红色荧光表达情况,设计引物f:atggcctcctccgagaacg(seqidno.:28),r:acgatggtgtagtcctcgttgt(seqidno.:29)扩增其mrna。以gadph为内参,设计引物gadph-f:gcaagttccacggcacag(seqidno.:30),gadph-r:cgccagtagaagcagggat(seqidno.:31)扩增内参序列。结果见转基因羊组织器官中均有红色荧光蛋白的转录,转基因羊扩增出0.64kb的红色荧光基因转录目的条带,而野生型未扩增出;同时,二者均扩增出内参基因0.47kb的gapdh内参条带(图11)。
采用caliper(珀金埃尔默公司)ivisluminaⅲ光学成像系统,对转基因羊各组织器官进行了荧光分析,并以正常羊做对照,结果见图12,在同样的荧光强度下,转基因羊各组织器官(小脑、大脑、延髓、耳、心、肝、脾、肺、肾、小肠,肌肉)中均有红色荧光表达,而对照野生型羊组织器官基本无红色,可见g6位点是一个可支持外源基因高效稳定表达的基因整合热点。
实施例5
含loxp序列的位点特异性重组载体ptg6-gfp的构建
设计g6位点特异性重组载体,以gfp为目的基因,以新霉素为筛选基因,在目的基因两侧连接两个含不同核心序列的loxp位点(lox66,lox2272),利用cre酶介导的位点特异性重组实现gfp在g6位点的基因替换。
以质粒pgfp-n3为模板,用含lox2272的引物及酶切位点的引物f8:ggggtaccataacttcgtataggatactt(seqidno.:32),r8:gaatgatatcgatctgacggttcactaaaccag(seqidno.:33)进行pcr,扩增642bp的含cmv基因启动子片段6。
以改造过的质粒pgfp-n3(删去了noti位点)为模板,设计含酶切位点的引物f9:gaatgatatcgccaccatggtgagcaaggg(seqidno.:34),r9:acgcgtcgacgatgagtttggacaaaccac(seqidno.:35)扩增952bp的gfp序列片段7。将片段6用kpni+ecorv酶切,片段7用ecorv+sali双酶切后,连入经kpni+sali双酶切的pbsk载体中,获得pbsk-gfp。
设计含lox71及酶切位点的引物f10:acgcgtcgacccaggcaggcagaagtatgc(seqidno.:36),r10:gaatgcggccgctaccgttcgtataatgtatgctatacgaagttatacagacatgataagatacattg(seqidno.:37),以质粒pcdna3为模板,扩增1509bp的neomycin序列片段8。将片段8用sali+noti双酶切后,连入经sali+noti双酶切的pbsk-gfp载体中,获得位点特异性重组载体ptg6-gfp(图13)。
实施例6
cre酶介导的羊体细胞中g6位点的基因替换
复苏g6-red-loxpg羊分离耳成纤维细胞,将其培养于含2mm的谷氨酰胺,1mm的丙酮酸钠,1×的非必需氨基酸,2ng/ml的碱性成纤维细胞生长因子,1000units/ml的小鼠白血病抑制因子,10%胎牛血清(gibco),100units/ml的青链霉素的glasgow基本培养基(gmem,gibco)中,细胞于37℃,5%co2培养箱中培养,传代二次后,取处于对数生长期的细胞准备进行基因转染及cre酶孵育。
将构建的重组载体ptg6-gfp转染(lipo3000,invitrogen)第3代的g6-red-loxpg山羊耳成纤维细胞(g6-red-loxpc),脂质体作用2小时后,加入终浓度为0.08mg/ml的细胞穿透性tat-cre孵育3小时,然后换成正常培养基继续培养。48小时后,将这些细胞消化后重新分到含0.8mg/mlg418(gibco)的选择性培养液中。大约8-9天后,用克隆环将其中生长良好且独立的细胞克隆挑出,转移到48孔板孔。然后传代至24孔,待24孔板长满后,收集2/3细胞冻存,1/3用于抽提基因组dna。共挑取独立、边缘清晰的细胞克隆25株,其中19个长势良好,收集细胞,采用天根基因组dna提取试剂盒抽提细胞基因组dna备用。
分别在g6位点5′端、3′端,重组载体内部设计两对引物检测经过位点特异性重组的细胞株,引物位置见图14,具体序列如下:f11:ttgtagaactggaatggtggcaa(seqidno.:38),r11:agttcaccttgatgcccgttctt(seqidno.:39);f12:atcaggacatagcgttggc(seqidno.:40),r12:acctcccttgctctggatgtagt(seqidno.:41)。
以挑取的细胞克隆基因组dna为模板,分别用两对引物f11r11及f12r12扩增发生位点特异性重组的细胞克隆基因组dna片段,扩增条件:95℃5min;95℃30s,56℃30s,72℃,1min,30cycles;72℃,10min。结果在16个检测细胞克隆中获得2个中靶阳性克隆,重组效率为12.5%,pcr检测结果见图(图15)。可见中靶克隆5’端扩出1.5kb条带,3’端扩出0.67kb条带,与预期相符,说明细胞克隆在g6位点发生了定位重组,绿色荧光蛋白表达框已定点插入了目标染色体位点,该细胞命名为g6-gfp-loxpc。将该细胞克隆置于荧光显微镜下镜检,见细胞发出稳定的绿色荧光(图16)。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!