一种氯唑沙宗的制备方法与流程
本发明涉及一种药物化合物氯唑沙宗(Chlorzoxazone)的制备方法,属于化学药物合成领域。
背景技术:
氯唑沙宗为一种中枢性骨骼肌松驰剂,用于治疗各种骨骼肌紊乱性疾病,如各种急慢性软组织(肌肉、韧带)扭伤、挫伤,运动后肌肉酸痛,肌肉、韧带、筋膜扭伤等,对中枢神经病变引起的肌肉痉挛以及慢性筋膜炎、儿童智力发育不良有一定疗效。
中国专利CN1560040A中,公开了一种氯唑沙宗的合成方法,主要合成路线如下所示:
该方法以2,5-二氯硝基苯为起始原料,经过还原、水解、环合三步反应,在还原反应过程中以LiAlH4或Raney/Ni为还原剂,以水、醇、醚或者四氢呋喃作溶剂得到还原产物,进行水解反应,以氢氧化钠、氢氧化钾、氰化钠、氰化钾、醇化钠或醇化钾为水解剂,以水、醚或芳香类为溶剂,得到水解产物,不经精制,进行下一步的环合反应,环合剂为脲素,产物即为氯唑沙宗,精致得到纯品。
该方法的优点是水解产物2-氨基-4-氯苯酚不需精制即可投入下一步反应,但还原产物2,5-二氯苯胺颜色较深,熔点低,易被氧化。且在进行水解反应的时候,由于邻位氨基的推电子作用,使得1位的氯在碱性条件下水解为羟基的难度加大,难以实现。
除上述合成方法外,何浩明等人在《中国医药工业杂志》Vol.23(11)489中还报道了一种2,5-二氯硝基苯经过在氢氧化钠水溶液中水解、以Na2S2为还原剂还原,继而在酸性条件下与脲素环合生成氯唑沙宗,经乙醇重结晶得到纯化的产品的路线。
此方法的优点在于原料易得,反应条件温和,但还原剂二硫化钠的使用会对环境造成污染,且产生相当多的固体废料,并且中间体2-氨基-4-氯苯酚容易被氧化变色,不易贮存,这都使其在工业化生产上造成困难。
技术实现要素:
本发明为了解决以上技术问题,提出了一种氯唑沙宗制备方法的制备方法。
本发明的目的通过以下技术方案来实现:
一种氯唑沙宗的制备方法,反应式如下所示,
具体包括如下步骤:
S1、合成4-氯-2-硝基苯酚;
S11、将原料2,5-二氯硝基苯在强碱水溶液中加热水解得到水解产物,所述水解加热到120~150℃,反应16~24小时;
S12、反应结束后冷却,中和,过滤,干燥得到纯度为≥99%的4-氯-2-硝基苯酚;
S2、催化合成:将S1所得的4-氯-2-硝基苯酚溶于甲醇,以Raney/Ni为催化剂,氢化还原生成2-氨基-4-氯苯酚;该中间产物直接进行环合反应,避免了分离纯化过程中被氧化而影响后续的反应纯度。
S3、过滤蒸馏:过滤除去Raney/Ni,滤液中加入适量的水以保持以下蒸馏时处于溶液状态,蒸馏除去甲醇,残留液即为2-氨基-4-氯苯酚的水溶液;
S4、环合反应:在残留液中加入活性炭脱色,并在酸性条件下以脲素为环合剂,升温至回流,发生环合反应生成氯唑沙宗;
S5、反应完成后冷却过滤得到粗品,粗品经过酸碱处理、脱色,得到氯唑沙宗纯品。
优选地,所述S11中所用的强碱为氢氧化钠、氢氧化钾中的一种,所述强碱水溶液的浓度为50~60g/L。
优选地,所述S12中采用浓盐酸进行中和至pH=2~5。
优选地,所述S3滤液中加水的量为滤液的两倍。
优选地,所述S4中的酸性条件为pH<6。
优选地,以上所述的一种氯唑沙宗的制备方法制得的氯唑沙宗,所述氯唑沙宗的纯度≥99%。
本发明方法的优点:整条合成路线一共两步,步骤二将还原反应和环合反应合为一步,不仅有效提高整体收率,而且缩短了反应周期。减少2-氨基-4-氯苯酚停留时间,避免了分离出的化合物被氧化变质。
采用本发明制得的氯唑沙宗颜色好纯度高稳定性好,经过简单重结晶即可得到纯品。收率达到76%以上,产品纯度达到99%以上。
步骤二中使用的还原催化剂Raney/Ni价格便宜,而且反应结束后可过滤回收反复循环使用,大大减少了环境污染,降低了生产成本,适合于工业化生产。
附图说明
图1是中间产物4-氯-2-硝基苯酚的核磁图谱-1HNMR示意图。
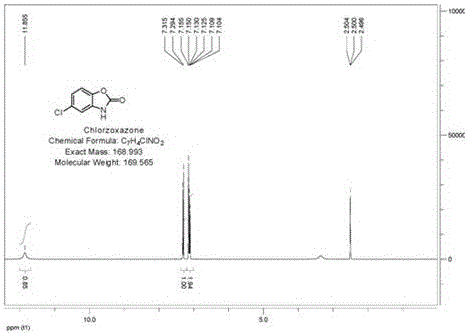
图2是氯唑沙宗的核磁图谱-1HNMR示意图。
具体实施方式
以下结合实施例具体说明本发明的制备方法,本发明的反应式如下所示,
中间产物的制备:4-氯-2-硝基苯酚(IV)的制备
在3L压力釜中加入1.6L 50~60g/L的NaOH水溶液,和106g 2,5-二氯硝基苯,密闭反应釜,开始搅拌,升温至120~150℃,通过色谱跟踪反应。
反应完毕,冷却至40℃~70℃,用浓盐酸中和至pH=2~5,冷却过滤,干燥得到淡黄色固体4-氯-2-硝基苯酚(IV),纯度为99.8%。
经核磁共振定性分析,检测数据如下:1H NMR(400MHz,DMSO-d6)δ7.15(d,1H),7.59(dd,1H),7.95(d,1H),11.30(br,1H)。检测图谱如图1所示。
以上述中间产物为进一步的原料进行氯唑沙宗的制备。
实施例一、氯唑沙宗(Chlorzoxazone)的制备
在350mL压力釜中加入50-100mL甲醇、1.0g Raney/Ni、10g 4-氯-2-硝基苯酚,密闭反应釜,开动搅拌,将反应体系中空气置换成氢气,且保持氢气压力0.1~0.2MPa,室温反应,色谱跟踪反应。
反应完毕,在氮气保护下,过滤除去Raney/Ni。向滤液中加入10-30mL水,蒸馏除去甲醇。向残留液中加入5-7mL 36%浓盐酸、0.5g活性炭,升温至50℃,搅拌半小时,过滤,用水洗涤。
向滤液中加入8.6g脲素,升温至回流反应1小时,补加浓盐酸,控制反应液为酸性,继续回流反应三小时,在此过程中,不停补加浓盐酸,确保反应液的酸性,之后,回流反应过夜。
反应完毕后,冷却,过滤得到褐色粗产品。将褐色粗品溶于1N氢氧化钠溶液,加入0.5g活性炭,升温至50℃,搅拌半小时,过滤。滤液用3N盐酸酸化至Ph=3~6,冷却过滤,干燥得到白色固体氯唑沙宗(Chlorzoxazone)7.4g,纯度为99%。
实施例二、氯唑沙宗(Chlorzoxazone)的制备
在2L压力釜中加入200-400mL甲醇、4.0g Raney/Ni、40g 4-氯-2-硝基苯酚,密闭反应釜,开动搅拌,将反应体系中空气置换成氢气,且保持氢气压力0.1~0.2MPa,室温反应。反应完毕,氮气保护,过滤除去Raney/Ni。向滤液中加入水40-120mL,蒸馏除去甲醇。向残留液中加入20-30mL 36%浓盐酸、2g活性炭,升温至50℃,搅拌半小时,过滤,用水洗涤。向滤液中加入34.4g脲素,升温至回流反应1小时,补加浓盐酸,控制反应液为酸性,继续回流反应三小时,在此过程中,不停补加浓盐酸,确保反应液的酸性,之后,回流反应过夜。反应完毕后,冷却,过滤得到褐色粗产品。将褐色粗品溶于1N 600mL氢氧化钠溶液,加入2g活性炭,升温至50℃,搅拌半小时,过滤。滤液用3N盐酸酸化至Ph=3~6,冷却过滤,干燥得到白色固体氯唑沙宗(Chlorzoxazone)29.7g,纯度大于99%。
经核磁共振定性分析,检测数据如下:1H NMR(400MHz,DMSO-d6)δ7.10~7.16(m,2H),7.30(d,1H),11.86(br,1H)。检测图谱如图2所示。
同时,通过依照中国药典2000年版二部附录IV A分光光度法测定,氯唑沙宗在244nm与288nm的波长处有最大吸收。
红外吸收图谱与对照的图谱(光谱集499图)一致。
本发明尚有多种具体的实施方式。凡采用等同替换或者等效变换而形成的所有技术方案,均落在本发明要求保护的范围之内。
- 还没有人留言评论。精彩留言会获得点赞!