一种氘代咪唑酮化合物的晶型及其制备方法和用途与流程
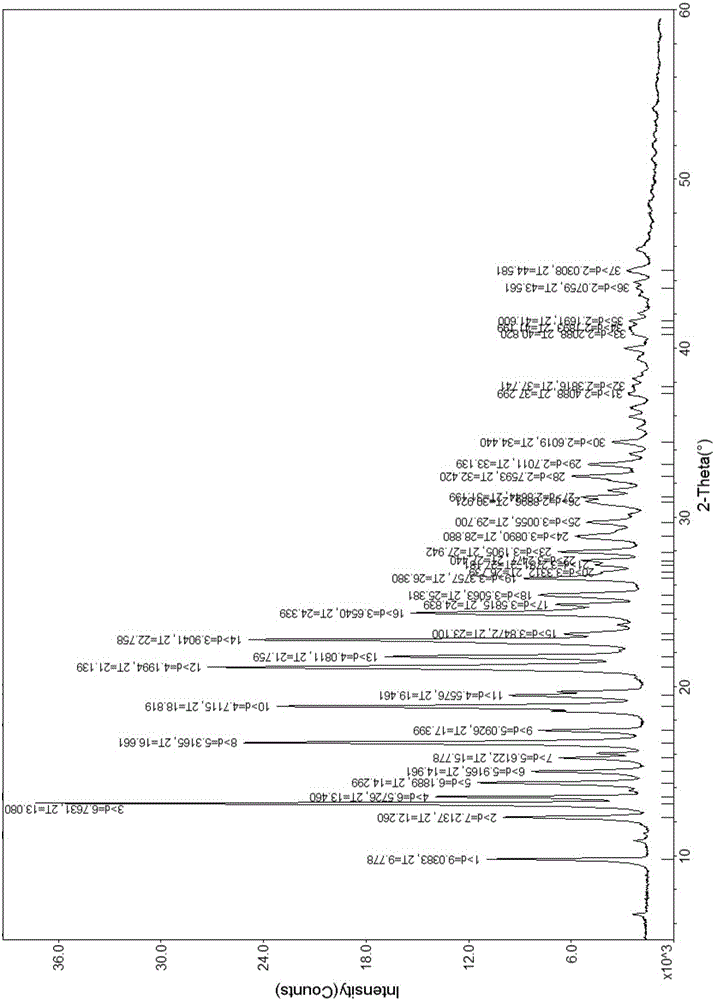
本发明涉及氘代咪唑酮化合物晶型Ⅰ及其制备方法和用途。
背景技术:
氘代咪唑酮化合物——4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺是具有以下化学结构的化合物:
专利CN201280052853已公开了其制备方法和用途,但是目前,并没有关于其晶型的报道。对于同一种化合物来说,通常会有两种或多种不同的结晶状态,而不同的晶型在稳定性上均有所差异。探索出稳定性更好的晶型,有利于药物的存储及运输,保障了药效的稳定了。
因此,对于药物而言,探索得到药物的多种晶型,从中寻找到稳定性优异的晶型品种具有非常重要的意义。
技术实现要素:
本发明提供了4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺新晶型I,该晶型稳定性好不易被氧化;且合成晶型过程中操作简单,可放大生产;在软胶囊内容物中溶解度好。
本发明提供了一种氘代咪唑酮化合物——4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的晶型Ⅰ,该晶型的X射线粉末衍射中,2θ衍射角度在12.3±0.2、13.1±0.2、15.0±0.2和17.5±0.2度处有特征峰。
进一步地,该晶型X射线粉末衍射中,2θ衍射角度还在9.8±0.2、13.5±0.2、14.3±0.2、16.7±0.2、18.9±0.2、21.1±0.2、21.8±0.2、22.8±0.2和24.4±0.2度处有特征峰。
更进一步地,该晶型X射线粉末衍射中,2θ衍射角度特征峰的相对强度值为:
其中,该晶型具有基本如图1或图2所示的X射线粉末衍射图谱。
其中,该晶型的熔点为192-209℃。
其中,该晶型具有基本如图3所示的DSC图谱。
本发明还提供了一种制备上述晶型Ⅰ的方法,包括以下步骤:
(1)将4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺溶解在溶剂中,所述溶剂不选自甲醇或甲酸中的任一种或其组合;
(2)结晶;
(3)分离出晶体,干燥即得。
进一步地,所述步骤(2)的结晶是搅拌冷却结晶;或加入抗溶剂,搅拌冷却结晶。
进一步地,在步骤(1)中,所述溶剂选自醇、腈、卤代烃、酰胺、亚砜、卤代烃、芳烃、酯、醚、酮、水、乙酸中的任一种或其组合。
进一步地,在步骤(1)中,所述溶剂选自乙醇、异丙醇、正丁醇、乙腈、N,N-二甲基甲酰胺、二甲基亚砜、甲苯、二甲苯、乙酸乙酯、乙酸丁酯、四氢呋喃、丙酮、甲基异丙基甲酮、甲基异丁基甲酮、水、乙酸中的任一种或其组合。
进一步地,在步骤(1)中,所述溶剂与4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的体积重量比为1~50:1mL/g。
进一步地,在步骤(1)的溶解过程中,温度为0℃至溶剂的回流温度。
进一步地,所述抗溶剂是水或C5-C10的烃类溶剂。
进一步地,在步骤(2)中,析晶温度为-20℃-100℃,优选的析晶的温度为3℃至室温。
本发明提供的制备上述晶型I的方法还包括:
a)提供HC-1119在溶剂中的溶液;以及b)分离HC-1119的晶型I。步骤a)中的提供溶液包括:
i)直接使用含有在HC-1119的合成过程中获得的HC-1119的反应混合物;或者
ii)将HC-1119溶解在溶剂中。
HC-1119的任何物理形式都可用于在步骤a)中提供HC-1119的溶液。任选地,当使用HC-1119的水合物时,在步骤a)之前或之后,可通过本领域中已知的技术例如蒸馏、加热、在合适的溶剂中制浆等来进行水减少或除去步骤。
在实施方案中,可将在HC-1119的合成过程中获得的HC-1119溶解在任何合适的溶剂中。此类合适的溶剂的实例包括但不限于:醇,例如C2-C6醇,例如乙醇、1-丙醇、2-丙醇(异丙醇)、1-丁醇、2-丁醇、叔丁醇;或腈,例如乙腈或丙腈;酰胺,例如N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮;亚砜,例如二甲基亚砜;卤代烃,例如二氯甲烷;芳烃,例如甲苯、二甲苯;酯,例如乙酸乙酯、乙酸正丙酯、乙酸正丁酯、乙酸异丙酯、乙酸异丁酯、乙酸叔丁酯;醚,例如乙醚、二异丙基醚、甲基叔丁基醚、四氢呋喃、1,4-二噁烷、2-甲氧基乙醇、苯甲醚;酮,例如丙酮、乙基甲基酮、二乙基酮、甲基异丁基酮;水;或一种或多种这些溶剂的任意混合物。
溶解温度可为约0℃至约溶剂的回流温度,或者低于约150℃,低于约130℃,低于约100℃,低于约70℃,低于约40℃,低于约20℃,低于约0℃,或者任何其它合适的温度,只要获得HC-1119的澄清溶液而不影响其质量。可以任选地用碳、煅烧(flux-calcined)硅藻土(Hyflow)或任何其它合适的材料处理所述溶液,以除去颜色、不溶性材料,改善溶液的澄清度,和/或除去可吸附于此类材料上的杂质。任选地,可将上述获得的溶液过滤以除去任何不溶性颗粒。可以通过过滤、离心、倾析或者在加压下或在减压下的任何其它合适的技术来适当地除去所述不溶性颗粒。可通过使所述溶液通过纸、玻璃纤维、布或其它膜材料,或者澄清剂(例如或Hyflow)床对其进行过滤。取决于所使用的设备以及溶液的浓度和温度,可能需要预热过滤装置以避免过早结晶。
步骤b)包括从在步骤a)中获得的溶液中分离HC-1119的晶型I。步骤b)中的分离HC-1119的晶型I通过包括冷却、速冷(crash cooling)、浓缩物质、加入抗溶剂、加入晶种以诱导结晶或蒸发等或其组合在内的方法进行。搅拌或其它替代方法例如振荡、搅动等也可以用于分离。或者,当形式R1的晶体存在于在步骤(a)中获得的溶液中时,这些晶体可以用作晶种,并且可以在步骤(b)中在此类形式R1的晶种存在下分离形式R1。
任选地,可以通过将合适的抗溶剂与在步骤a)中获得的溶液组合来进行分离。如本文所用,抗溶剂是指HC-1119在其中微溶或难溶的液体。抗溶剂对HC-1119的质量没有不利的影响,并且它可以帮助溶解的原料凝固或沉淀。可使用的合适的抗溶剂包括但不限于:水;饱和或不饱和的直链或支链的、环状或非环状的C1-C10烃,例如己烷、庚烷、环己烷或甲基环己烷;醚,例如乙醚、二异丙基醚、四氢呋喃、二噁烷或二甲氧基乙烷;或其混合物。
合适的分离温度可为低于约100℃,低于约80℃,低于约60℃,低于约40℃,低于约20℃,低于约10℃,低于约5℃,低于约0℃,低于约-10℃,低于约-20℃,或者任何其它合适的温度。
分离的HC-1119的晶型I可以通过包括倾析、离心、蒸发、重力过滤、抽滤或者在加压下或在减压下的任何其它用于固体回收的技术在内的方法进行回收。回收的固体可以任选地进行干燥。所述干燥可以在盘式干燥器、真空烘箱、空气烘箱、锥形真空干燥器(conevacuum dryer)、旋转式真空干燥器、流化床干燥器、旋转闪蒸干燥器、快速干燥器等中进行。所述干燥可以在低于约100℃、低于约80℃、低于约60℃、低于约50℃、低于约30℃的温度或任何其它合适的温度下,在大气压或减压下进行,只要HC-1119的质量不劣化。所述干燥可以进行任何期望的次数,直到实现所需的产物质量。干燥的产物可以任选地经历粉碎(size reduction)操作,以产生期望的粒度。可在产物的干燥前或干燥完成后进行研磨或微粉化。可用于减小粒度的技术包括但不限于球磨、辊磨和锤磨,以及喷射研磨(jetmilling)。
本发明还提供了一种药物组合物,它是由上述的4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的晶型Ⅰ作为活性成分,加上药学上可接受的辅料制备而成的制剂。
进一步地,本发明提供了上述4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的晶型Ⅰ在制备雄性激素受体拮抗剂,或制备治疗或/和预防雄性激素受体活性相关疾病的药物中的应用。
其中,所述疾病选自但不局限于脱发、再生发、暗疮、青春痘或前列腺癌。
除此之外,在对化合物HC-1119的晶型筛选中,发明人还发现了其晶型Ⅱ,该晶型的X射线粉末衍射中,2θ衍射角度在4.8±0.2、11.3±0.2和20.2±0.2度处有特征峰。
进一步地,该晶型X射线粉末衍射中,2θ衍射角度还在9.7±0.2、14.5±0.2、15.6±0.2、16.9±0.2和25.5±0.2度处有特征峰。
进一步地,该晶型X射线粉末衍射中,2θ衍射角度特征峰的相对强度值为:
其中,该晶型具有基本如图4或图5所示的X射线粉末衍射图谱。
其中,该晶型的熔点为114-139℃。
其中,该晶型具有基本如图6所示的DSC图谱。
本发明还提供了一种制备上述晶型Ⅱ的方法,它包括以下步骤:
(1)将4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺溶解在溶剂中,所述溶剂选自甲醇或甲酸中的任一种或其组合;
(2)结晶;
(3)分离出晶体,干燥即得。
进一步地,所述步骤(2)的结晶是搅拌冷却结晶;或加入抗溶剂,搅拌冷却结晶。
进一步地,在步骤(1)中,所述溶剂与4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的体积重量比为2.5~10:1mL/g。
进一步地,在步骤(1)的溶解过程中,温度为0℃至溶剂的回流温度。
进一步地,所述抗溶剂是水或C5-C10的烃类溶剂。
进一步地,在步骤(2)中,析晶温度为-20℃-100℃,优选的析晶的温度为3℃至室温。
本发明提供的制备上述晶型Ⅱ的方法还包括:a)提供HC-1119在甲醇或甲酸中的溶液;以及b)分离HC-1119的晶型II。
步骤a)中的提供溶液包括:
i)直接使用含有在HC-1119的合成过程中获得的HC-1119的反应混合物;或者
ii)将HC-1119溶解在甲醇或甲酸中。
HC-1119的任何物理形式都可以用于在步骤a)中提供HC-1119的溶液。任选地,当使用HC-1119的水合物时,在步骤a)之前或之后,可通过本领域中已知的技术例如蒸馏、加热、在合适的溶剂中制浆等进行水减少或除去步骤。
在实施方案中,可将在HC-1119的合成过程中获得的HC-1119溶解在甲醇或甲酸或其任意混合物中。
溶解温度可为约0℃至约溶剂的回流温度,或者低于约100℃,低于约80℃,低于约60℃,低于约40℃,低于约20℃,低于约10℃,或者任何其它合适的温度,只要获得HC-1119的澄清溶液而不影响其质量。可以任选地用碳、煅烧硅藻土(Hyflow)或任何其它合适的材料处理所述溶液,以除去颜色、不溶性材料,改善溶液的澄清度,和/或除去可吸附于此类材料上的杂质。任选地,可将上述获得的溶液过滤以除去任何不溶性颗粒。可以通过过滤、离心、倾析或者在加压下或在减压下的任何其它合适的技术来适当地除去所述不溶性颗粒。可通过使所述溶液通过纸、玻璃纤维、布或其它膜材料,或者澄清剂(例如或Hyflow)床对其进行过滤。取决于所使用的设备以及溶液的浓度和温度,可能需要预热过滤装置以避免过早结晶。
步骤b)包括从在步骤a)中获得的溶液中分离HC-1119的晶型II。步骤b)中的分离HC-1119的晶型II可以涉及包括冷却、浓缩物质、加入抗溶剂、加入晶种以诱导结晶等在内的方法。搅拌或其它替代方法例如振荡、搅动等也可以用于分离。
任选地,可以通过将合适的抗溶剂与在步骤a)中获得的溶液组合来进行分离。如本文所用,抗溶剂是指HC-1119在其中微溶或难溶的液体。抗溶剂对HC-1119的质量没有不利的影响,并且它可以帮助溶解的原料凝固或沉淀。可使用的合适的抗溶剂包括但不限于:饱和或不饱和的直链或支链的、环状或非环状的C1-C10烃,例如己烷、庚烷、环己烷或甲基环己烷;醚,例如乙醚、二异丙基醚、四氢呋喃、二噁烷或二甲氧基乙烷;或其任意混合物。
合适的分离温度可为低于约100℃,低于约80℃,低于约60℃,低于约40℃,低于约20℃,低于约10℃,低于约5℃,低于约0℃,低于约-10℃,低于约-20℃,或者任何其它合适的温度。
分离的HC-1119的晶型II可以通过包括倾析、离心、重力过滤、抽滤或者在加压下或在减压下的任何其它用于固体回收的技术在内的方法进行回收。回收的固体可以任选地进行干燥。所述干燥可以在盘式干燥器、真空烘箱、空气烘箱、锥形真空干燥器、旋转式真空干燥器、流化床干燥器、旋转闪蒸干燥器、快速干燥器等中进行。所述干燥可以在低于约100℃、低于约80℃、低于约60℃、低于约50℃、低于约30℃的温度或任何其它合适的温度下,在大气压或减压下进行,只要HC-1119的质量不劣化。所述干燥可以进行任何期望的次数,直到实现所需的产物质量。干燥的产物可以任选地经历粉碎操作,以产生期望的粒度。可在产物的干燥前或干燥完成后进行研磨或微粉化。可用于减小粒度的技术包括但不限于球磨、辊磨和锤磨,以及喷射研磨。
本发明还提供了一种药物组合物,它是由上述4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的晶型Ⅱ作为活性成分,加上药学上可接受的辅料制备而成的制剂。
进一步地,本发明还提供了上述的4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的晶型Ⅱ在制备雄性激素受体拮抗剂,或制备治疗或/和预防雄性激素受体活性相关疾病的药物中的应用。
其中,所述疾病选自脱发、再生发、暗疮、青春痘或前列腺癌。
发明人重复专利CN201280052853得到的化合物,其晶型为无定形。其中,该无定形化合物具有基本如图7或图8所示的X射线粉末衍射图谱。
本发明还提供了一种制备上述无定形化合物的方法,它包括以下步骤:
(1)将4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺溶解在溶剂中;
(2)冷却或不冷却;
(3)快速除去溶剂或快速析出固体并分离,即得。
进一步地,在步骤(1)中,所述溶剂选自醇、腈、卤代烃、酰胺、亚砜、卤代烃、芳烃、酯、醚、酮、水、乙酸中的任一种或其组合。
进一步地,在步骤(1)中,所述溶剂选自甲醇、乙醇、异丙醇、正丁醇、乙腈、N,N-二甲基甲酰胺、二甲基亚砜、甲苯、二甲苯、乙酸乙酯、乙酸丁酯、四氢呋喃、丙酮、甲基异丙基甲酮、甲基异丁基甲酮、水、乙酸中的任一种或其组合。
进一步地,在步骤(1)中,所述溶剂与4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的体积重量比为2.5~10:1mL/g。
进一步地,在步骤(1)的溶解过程中,温度为0℃至溶剂的回流温度。
进一步地,在步骤(2)中,析晶温度为-20℃-100℃,优选的析晶的温度为3℃至室温。
进一步地,在步骤(3)中,所述快速除去溶剂指的是1分钟内至少除去10mL溶剂,所述溶剂中至少溶解有1g 4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺。
进一步地,所述步骤(3)的除去溶剂是通过旋转蒸发进行的。
进一步地,所述步骤(3)的快速析出固体是通过加入抗溶剂的进行的。
进一步地,所述抗溶剂是水或C5-C10的烃类溶剂。
本发明提供的制备上述无定形化合物的方法还包括:
a)提供HC-1119在溶剂中的溶液;以及b)分离HC-1119的无定形化合物。
步骤a)中的提供HC-1119的溶液包括:
i)直接使用含有在HC-1119的合成过程中获得的HC-1119的反应混合物;或者
ii)将HC-1119溶解在溶剂中。
HC-1119的任何物理形式都可以用于在步骤a)中提供HC-1119的溶液。溶解温度可为约0℃至约溶剂的回流温度,或者低于约60℃,低于约50℃,低于约40℃,低于约30℃,低于约20℃,低于约10℃,或者任何其它合适的温度,只要获得HC-1119的澄清溶液而不影响其质量。可以任选地用碳、煅烧硅藻土(Hyflow)或任何其它合适的材料处理所述溶液,以除去颜色、不溶性材料,改善溶液的澄清度,和/或除去可吸附于此类材料上的杂质。任选地,可将上述获得的溶液过滤以除去任何不溶性颗粒。可以通过过滤、离心、倾析或者在加压下或在减压下的任何其它合适的技术来适当地除去所述不溶性颗粒。可通过使所述溶液通过纸、玻璃纤维、布或其它膜材料,或者澄清剂(例如或Hyflow)床对其进行过滤。取决于所使用的设备以及溶液的浓度和温度,可能需要预热过滤装置以避免过早结晶。
在实施方案中,可将HC-1119溶解在任何合适的溶剂中。合适的溶剂包括对化合物没有不利的影响并且可以将原料溶解到有用的程度的任何溶剂。此类溶剂的实例包括但不限于:醚,例如乙醚、二异丙基醚、四氢呋喃、二噁烷或二甲氧基乙烷;酮,例如丙酮、甲基乙基酮、甲基异丁基酮或二乙基酮;酯,例如乙酸乙酯、乙酸丙酯、乙酸异丙酯或乙酸丁酯;醇,例如甲醇、乙醇、1-丙醇、2-丙醇(异丙醇)、2-甲氧基乙醇、1-丁醇、2-丁醇、异丁醇、叔丁醇、2-乙氧基乙醇、二乙二醇、1-戊醇、2-戊醇或3-戊醇、新戊醇、叔戊醇、二乙二醇单甲醚、环己醇、甘油或C1-C6醇;腈,例如乙腈或丙腈;酰胺,例如甲酰胺、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮或六甲基磷酸三酰胺;亚砜,例如二甲基亚砜;卤代烃,例如二氯甲烷、氯仿、四氯化碳或氯苯;芳烃,例如甲苯;或其两种或更多种的任意混合物。
步骤b)包括从在步骤a)中获得的溶液中分离HC-1119的无定形化合物。步骤b)中的分离HC-1119的无定形化合物可以涉及包括除去溶剂、冷却、速冷、浓缩物质、蒸发、快速蒸发、简单蒸发、旋转干燥、喷雾干燥、薄膜干燥、搅动吸滤器干燥、加压吸滤器干燥、冷冻干燥、加入抗溶剂、用溶剂萃取等在内的方法。搅拌或其它替代方法例如振荡、搅动等也可以用于分离。所分离的HC-1119的无定形化合物可能携带一定量的包藏的母液,并且可能具有高于期望水平的杂质。如果需要,可以用溶剂或溶剂混合物洗涤这种无定形化合物以洗出杂质。
制备无定形化合物的关键在于在较短的时间内除去溶剂或是析出固体,以避免化合物形成晶型,可以通过本领域常用的制备无定形化合物的方法得到。
用旋转蒸发制备无定形化合物的关键在于,形成HC-1119在可溶溶剂中的溶液,低温(温度度不超过50℃),以至少5毫升/分钟的速率去除溶剂,优选5-15毫升/分钟。
合适的分离温度可为低于约120℃,低于约80℃,低于约60℃,低于约40℃,低于约30℃,低于约20℃,低于约10℃,低于约0℃,低于约-10℃,低于约-40℃,或者任何其它合适的温度。
任选地,可以通过将合适的抗溶剂与在步骤a)中获得的溶液组合来进行分离。如本文所用,抗溶剂是指HC-1119在其中微溶或难溶的液体。惰性抗溶剂对反应没有不利的影响,并且它可以帮助溶解的原料凝固或沉淀。可使用的合适的抗溶剂包括但不限于:饱和或不饱和的直链或支链的、环状或非环状的C1-C10烃,例如庚烷、环己烷或甲基环己烷;水;或其任意混合物。
回收的固体可以任选地进行干燥。所述干燥可以在盘式干燥器、真空烘箱、空气烘箱、锥形真空干燥器、旋转式真空干燥器、流化床干燥器、旋转闪蒸干燥器、快速干燥器等中进行。所述干燥可以在低于约100℃、低于约80℃、低于约60℃、低于约50℃、低于约30℃的温度或任何其它合适的温度下,在大气压或减压下进行,只要HC-1119的质量不劣化。所述干燥可以进行任何期望的次数,直到实现所需的产物质量。干燥的产物可以任选地经历粉碎操作,以产生期望的粒度。可在产物的干燥前或干燥完成后进行研磨或微粉化。可用于减小粒度的技术包括但不限于球磨、辊磨或锤磨,或者喷射研磨。
本发明还提供了一种氘代咪唑酮化合物——4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的无定形化合物,该无定形化合物是以4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺为原料,通过上述制备无定形化合物的方法制备得到的或是本领域常规的制备无定形化合物的方法得到的。
本发明还提供了一种药物组合物,它是由上述的4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的无定形化合物作为活性成分,加上药学上可接受的辅料制备而成的制剂。
进一步地,本发明还提供了上述的4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-硫代-1-咪唑烷基}-2-氟-N-三氘代甲基苯酰胺的无定形化合物在制备雄性激素受体拮抗剂,或制备治疗或/和预防雄性激素受体活性相关疾病的药物中的应用。
其中,所述疾病选自脱发、再生发、暗疮、青春痘或前列腺癌。
本发明中,C5-C10的烃类溶剂指的是C5、C6、C7、C8、C9、C10的烃类溶剂,即具有5-10个碳原子的烷烃、烯烃、炔烃、环烃或芳香烃溶剂,例如正己烷、环己烷、苯、甲苯等等。
稳定性试验证明,本发明的晶型Ⅰ,晶型稳定,重现性好,放置稳定,显著优于现有的无定形物以及新发现的晶型Ⅱ,使得HC-1119原料药在存储期内质量更稳定。同时,本发明的晶型Ⅰ,制备工艺简单,成本低,具有广阔的市场前景。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
图1为通过实施例1的操作获得的晶型I的PXRD(X射线粉末衍射)图形。
图2为通过实施例12的操作获得的晶型I的PXRD图形。
图3为通过实施例1的操作获得的晶型I的DSC图谱。
图4为通过实施例18的操作获得的晶型II的PXRD图形。
图5为通过实施例19的操作获得的晶型II的PXRD图形。
图6为通过实施例4的操作获得的晶型I的DSC图谱。
图7为通过实施例20的操作获得的无定形化合物的PXRD图形。
图8为通过实施例21的操作获得的无定形化合物的PXRD图形。
具体实施方式
实施例1本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加10mL无水乙醇加热至70℃。加热至完全溶清后,继续搅拌10分钟,冷却至室温,再冰-水浴冷至3℃,搅拌10分钟过滤,滤饼用1mL冰无水乙醇淋洗,50℃真空干燥,得晶型I。
实施例2本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加11mL混合溶剂(DMSO:乙酸异丙酯=1:10),搅拌至完全溶清,加15%食盐水萃洗两次,每次6mL。有机相用无水硫酸钠干燥,过滤,50℃旋出溶剂,得到油状物,加10mL异丙醇打浆,过夜,过滤。50℃真空干燥得到晶型I。
实施例3本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加10mL乙腈,搅拌至完全溶清,滴加到装有30mL水的50mL单口瓶中,搅拌3小时,过滤,50℃真空干燥,得到晶型I。
实施例4本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加20mL异丙醇加热至70℃。加热至完全溶清后,继续搅拌10分钟,冷却至室温,再冰-水浴冷至3℃,搅拌30分钟过滤,滤饼用1mL冰异丙醇淋洗,50℃真空干燥,得到晶型I。
实施例5本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加10mL乙酸,室温搅拌30分钟至完全溶清,滴加20mL水,搅拌30分钟过滤,50℃真空干燥,得到晶型I。
实施例6本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加5mL乙酸乙酯,加热至50℃。加热至完全溶清后,继续搅拌10分钟,冰-水浴冷至5℃,搅拌3小时过滤,50℃真空干燥,得到晶型I。
实施例7本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加10mL乙酸丁酯,加热至70℃。加热至完全溶清后,继续搅拌10分钟,冰-水浴冷至3℃,搅拌10分钟过滤,50℃真空干燥,得到晶型I。
实施例8本发明晶型Ⅰ的制备
取2.0g HC-1119固体,加2.5mL四氢呋喃,加热至60℃。加热至完全溶清后,继续搅拌20分钟,冰-水浴冷至3℃,搅拌3小时过滤,50℃真空干燥,得到晶型I。
实施例9本发明晶型Ⅰ的制备
取2.0g HC-1119固体,加10mL甲基异丙基甲酮,加热至50℃。加热至完全溶清后,继续搅拌20分钟,冰-水浴冷至3℃,搅拌30分钟过滤,50℃真空干燥,得到晶型I。
实施例10本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加50mL甲苯,加热至90℃。加热至完全溶清后,继续搅拌20分钟,冰-水浴冷至3℃,搅拌30分钟过滤,50℃真空干燥,得到晶型I。
实施例11本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加10mL乙醇,加热至65℃。加热至完全溶清后,滴加20mL正庚烷,冰-水浴冷至3℃,搅拌20分钟过滤,50℃真空干燥,得到晶型I。
实施例12本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加20mL正丁醇,加热至90℃。加热至完全溶清后,继续搅拌20分钟,冰-水浴冷至3℃,搅拌15分钟过滤,50℃真空干燥,得到晶型I。
实施例13本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加50mL二甲苯,加热至100℃。加热至完全溶清后,继续搅拌20分钟,冰-水浴冷至3℃,搅拌15分钟过滤,50℃真空干燥,得到晶型I。
实施例14本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加25mL丙酮,25mL水,加热至50℃。加热至完全溶清后,继续搅拌10分钟,冰-水浴冷至3℃,搅拌1小时过滤,50℃真空干燥,得到晶型I。
实施例15本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加5mLDMSO,室温搅拌30分钟至完全溶清,滴加20mL水,搅拌2小时过滤,50℃真空干燥,得到晶型I。
实施例16本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加5mL甲基异丁基甲酮,加热至70℃。加热至完全溶清后,继续搅拌30分钟,冰-水浴冷至3℃,搅拌4小时过滤,50℃真空干燥,得到晶型I。
实施例17本发明晶型Ⅰ的制备
取1.0g HC-1119固体,加5mLDMF,室温搅拌30分钟至完全溶清,滴加20mL水,搅拌2小时过滤,50℃真空干燥,得到晶型I。
实施例18晶型Ⅱ的制备
取1.0g HC-1119固体,加10mL无水甲醇加热至60℃。加热至完全溶清后,继续搅拌10分钟,冷却至室温,再冰-水浴冷至3℃,搅拌10分钟过滤,滤饼用1mL冰无水甲醇淋洗,50℃真空干燥,得晶型II。
实施例19晶型Ⅱ的制备
取2.0g HC-1119固体,加5mL甲酸,加热至70℃。加热至完全溶清后,继续搅拌10分钟,冷却至室温,再冰-水浴冷至3℃,搅拌30分钟过滤,50℃真空干燥,得到晶型II。实施例20无定形化合物的制备
取1.0g HC-1119固体,加10mL丙酮,超声至完全溶清。35℃旋干,去除溶剂的速率约为10mL/分钟,得无定形。
实施例21无定形化合物的制备
取2.0g HC-1119固体,加5mL乙腈,加热至50℃。加热至完全溶清后,冰-水冷至室温,40℃旋干,得到无定形。
以下通过实验例具体说明本发明的有益效果。
实验例1长期稳定性实验
取实施例1制备的HC-1119晶型Ⅰ、实施例18制备的HC-1119晶型Ⅱ和重复专利CN201280052853得到的无定形物,分别用铝塑复合膜袋进行封装,于40℃±2℃,相对湿度75%±5%的条件下,于1、3月、6月、9月、12月月末取样检测,结果如下表1所示。
表1晶型的长期稳定性实验结果(0-12月)
由上述数据可以看出,HC-1119晶型I,晶型稳定,重现性好,放置稳定,使得HC-1119原料药在存储期内质量更稳定。此外,6月、9月和12月末用显微镜法进行晶型检查,结果本发明晶型Ⅰ无明显变化,而无定形物逐渐向固体颗粒状晶型转变。
综上所述,本发明的晶型Ⅰ,晶型稳定,重现性好,放置稳定,显著优于现有的无定形物以及新发现的晶型Ⅱ,使得HC-1119原料药在存储期内质量更稳定。同时,本发明的晶型Ⅰ,制备工艺简单,成本低,具有广阔的市场前景。
- 还没有人留言评论。精彩留言会获得点赞!