一种利用高效液相色谱法拆分R/S‑氯卡色林的方法与流程
本发明属于分析化学领域,涉及一种利用高效液相色谱法拆分R/S-氯卡色林的方法。
背景技术:
氯卡色林是一种新型减肥药,于2012年6月27日被美国食品药品管理局(FDA)批准上市。其英文名是Lorcaserin,化学名为8-氯-1-甲基-2,3,4,5-四氢-1H-3-苯并氮杂卓(8-chloro-1-methyl-2,3,4,5-tetrahydro-1H-3-benzazepin),分子式为C11H15Cl2N·0.5H2O,相对分子质量为195.69,结构式为:
氯卡色林一种能起到调节食欲和减少食物摄取作用的高选择性的5-羟色胺2C受体激动剂,通过5-HT2C受体的刺激来增加CNS血清素活性,激活POMC系统,释放黑皮质激素来调节热量平衡,从而影响下行途径,促进分解代谢和合成代谢抑制,有效控制体重。该药获准用于成人体质指数(BMI)≥27的肥胖或超重者,并且患者至少有一项与体重相关的疾病(如高血压、2型糖尿病或高脂血症)。
手性药物是指药物分子结构中引入手性中心后,得到的一对互为实物与镜像的对映异构体。这些对映异构体的理化性质基本相似,仅仅是旋光性有所差别,分别被命名为R-型(右旋)或S-型(左旋)、外消旋。绝大多数的药物由手性分子构成,两种手性分子可能具有明显不同的生物活性。药物分子必须与受体(起反应的物质)分子几何结构匹配,才能起到应有的药效,多数情况下只有一种药物对映体有显著的药理活性,而另一种对映体活性较低或没有活性,甚至具有毒副效应。
8-氯-1-甲基-2,3,4,5-四氢-1H-3-苯并氮杂卓分子为R/S外消旋体混合物,需进一步拆分为具有生物活性的R构型氯卡色林。在合成氯卡色林的过程中,S构型可能会由于去除不完全,从而影响药物纯度和质量。对于氯卡色林合成过程中引入的S构型,在原料药中是需要进行质量控制的。在手性药物对映体分离研究中,高效液相色谱法已成为使用最广泛和有效的方法之一。
由于绿卡色林分子小,其S构型和R构型在空间构象上的区分度不大,因此一般的手性柱很难拆分,需要找到一种刚好和其手性构型匹配一致的色谱固定相才能将其有效分离。目前为止还没有相关文献发表过绿卡色林手性分离的方法。
因此,实现氯卡色林对映体的有效分离,在氯卡色林原料药质量控制方面具有重要的现实意义。实验中我们主要对氯卡色林对映体的有效拆分的方法进行了研究。
技术实现要素:
本发明的目的在于提供一种利用高效液相色谱法拆分R/S-氯卡色林的方法。该方法能够高效、灵敏地实现氯卡色林对映体的检测和分离,从而保证氯卡色林R构型的纯度,可用于其终产品原料药的质量控制。
本发明的目的是通过下列技术方案实现的:
一种拆分R/S-氯卡色林对映体的方法,该方法采用高效液相色谱法,以直链淀粉类手性柱为色谱柱,以正已烷、醇和碱性添加剂为正相流动相。
所述的方法,其中色谱柱为Chiralpak IF手性柱,以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相。
所述的方法,其中流动相中正已烷、醇和碱性添加剂的体积百分比为49.6-98.9%:1-50%:0.1-0.4%。
所述的方法,其中正相流动相中醇为乙醇或异丙醇,碱性添加剂为二乙胺。
所述的方法,其中色谱柱为Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm),以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相,正相流动相中正已烷、乙醇和二乙胺的体积百分比为49.6-98.9%:1-50%:0.1-0.4%,流动相流速为0.4~1.0mL/min,紫外检测,检测波长为260-290nm,色谱柱柱温为5~40℃,进样量为10μL。
所述的方法,其中正相流动相中正已烷、乙醇和二乙胺的体积百分比为74.7%:25%:0.3%。
所述的方法,其中流动相流速为0.8mL/min。
所述的方法,其中色谱柱柱温为35℃。
所述的方法,其中待测样品采用甲醇溶液溶解,正己烷稀释。
所述的方法,其中所用的高效液相色谱仪为:安捷伦1260高效液相色谱仪(Agilent LC 1260)。配置有G1311的1260Quat泵(G1311 1260Quat pump VL)、G1316A恒温柱箱、G1329B自动液体进样器进样器(G1329B 1260ALS)、G4212B二极管阵列检测器(G4212B 1260DAD)。
具体地说,该方法包括下列步骤:
(1)待测样品预处理:取R-氯卡色林和氯卡色林消旋体各4mg,分别用1mL甲醇溶液溶解后,用正己烷稀释10倍,配制成浓度为400μg/mL的溶液,再取0.4mLR-氯卡色林溶液和0.8mL氯卡色林消旋体溶液混合后得到样品溶液,此时溶液中R-氯卡色林和S-氯卡色林的含量比为2:1,则R-氯卡色林和S-氯卡色林的色谱峰峰面积之比应为2:1;
(2)利用高效液相色谱仪,以直链淀粉类手性柱为色谱柱,以正己烷、醇和碱性添加剂为正相流动相,其中所述的醇为乙醇或异丙醇,优选乙醇;所述的碱性添加剂为二乙胺。控制流动相流速为0.4~1.0mL/min,优选0.8mL/min,检测波长为260-290nm,优选270nm,色谱柱柱温箱温度为5~40℃,优选35℃,进样量为10μL,将上述R/S-氯卡色林样品溶液进行手性分离;
上述方法中,由正己烷、醇和碱性添加剂组成的正相流动相中,按体积百分比计算,即正己烷:醇:碱性添加剂为49.6-98.9%;1-50%;0.1-0.4%,合计100%。
上述方法中,所用的直链淀粉类手性柱为Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm),以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相。
上述方法中,高效液相色谱仪的型号,无特别要求,本发明所用的高效液相色谱仪为:安捷伦1260高效液相色谱仪(Agilent LC1260),采用其他高效液相色谱仪也可以。
本发明的有益效果:
本发明的一种利用高效液相色谱法拆分R/S-氯卡色林的方法,使用Chiralpak IF手性色谱柱,并且使用正己烷、醇、碱性添加剂组成正相流动相,对R/S-氯卡色林分离度较好,达到完全的基线分离;采用的碱性添加剂二乙胺能够有效的改善峰形,达到分好的分离效果,本发明解决了R/S-氯卡色林的检测和分离问题,可用于氯卡色林原料药的质量控制。如果需要定量检测可通过与标准品的HPLC图对照即可算出。
附图说明
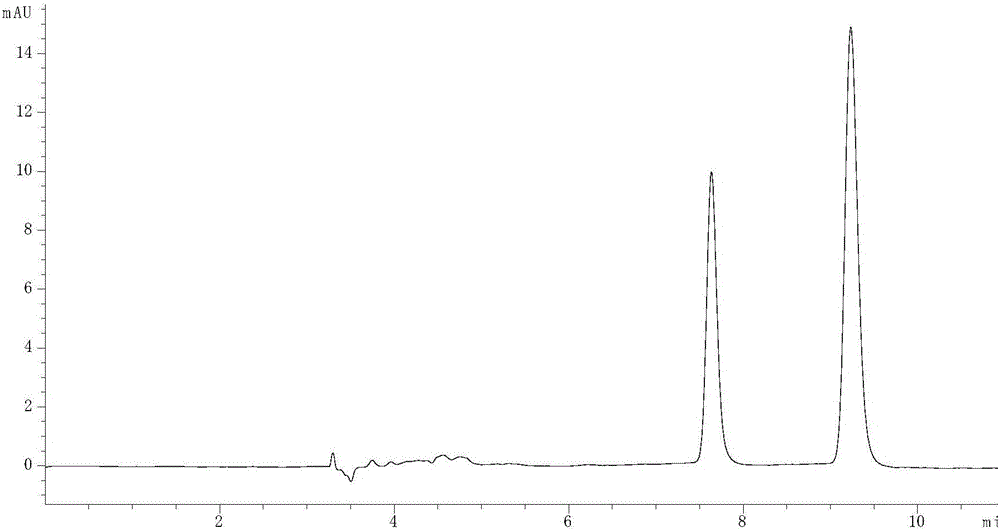
图1为实施例1时氯卡色林对映体HPLC图,色谱条件为:色谱柱:Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm);正己烷:乙醇:二乙胺=74.7:25:0.3(v/v/v);流速为0.8mL/min;检测波长为270nm;色谱柱柱温箱温度为35℃;进样量为10μL。
图2A为实施例2时氯卡色林对映体HPLC图,色谱条件为:色谱柱:Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm);正己烷:乙醇:二乙胺=74.7:25:0.3(v/v/v);流速为0.8mL/min;检测波长为270nm;色谱柱柱温箱温度为35℃;进样量为10μL。
图2B为实施例2时氯卡色林对映体HPLC图,色谱条件除流速为0.6mL/min外与图2A的条件相同。
图2C为实施例2时氯卡色林对映体HPLC图,色谱条件除流速为0.4mL/min外与图2A的条件相同。
图3为实施例3时氯卡色林对映体HPLC图,色谱条件为:色谱柱:Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm);正己烷:异丙醇:二乙胺=84.7:15:0.3(v/v/v);流速为0.8mL/min;检测波长为270nm;色谱柱柱温箱温度为35℃;进样量为10μL。
图4为实施例3时氯卡色林对映体HPLC图,色谱条件为:色谱柱:Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm);正己烷:乙醇:二乙胺=84.7:15:0.3(v/v/v);流速为0.8mL/min;检测波长为270nm;色谱柱柱温箱温度为35℃;进样量为10μL。
图5为采用OD-RH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:KPF6(100mM):CH3OH=60:40,流速为0.5mL,温度为25℃,进样为用乙腈配制的浓度为0.3mg/L的5μL的外消旋体。
图6为采用OD-RH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:NH4HCO3(20mM,pH8.3)/ACN=60/40,流速0.4ml/min,25℃,进样为用乙腈配制的浓度为0.3mg/L的5uL的外消旋体。
图7为采用OD-RH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:NH4Ac(10mM,pH7.0)/ACN=60/40,流速0.2ml/min,25℃,进样为用乙腈配制的浓度为0.3mg/L的5μL的外消旋体。
图8为采用ADH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:异丙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
图9为采用ASH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:异丙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
图10为采用ODH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:异丙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
图11为采用OJH柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:异丙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
图12为采用IA-H柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:乙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
图13为采用IB-H柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:乙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
图14为采用IC-H柱进行氯卡色林对映体分离的高效液相色谱图。色谱条件:正己烷:乙醇:二乙胺为80%:19.8%:0.2%。流速0.8mL/min,检测波长为270nm,进样为实施例4(1)中配制的旋光异构体混合物,进样量为10μL。
具体实施方式:
以下通过实施形式,对本发明涉及的氯卡色林对映体的分离检测方法作进一步的详细说明,但不应将此理解为本发明上述主题的范围仅限于以下的实例,凡基于本发明上述内容所实现的技术均属于本发明的范围。
实施例中采用的仪器及试剂:
直链淀粉类手性柱采用250mm×4.6mm I.D.粒度5μm,Chiralpak IF柱(大赛璐药物手性技术(上海)Daicel Chiral Technologies China,Shanghai),其硅胶表面共价键合有直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)。
正己烷、异丙醇、乙醇均为色谱纯试剂(TEDIA,美国天地高纯溶剂)。
二乙胺为分析纯试剂(上海凌峰化学试剂有限公司)。
安捷伦1260高效液相色谱仪,配置有G1311的1260Quat泵、G1316A恒温柱箱、G1329B自动液体进样器进样器、G4212B二极管阵列检测器。
实施例1:二乙胺对分离效果的影响
(1)取R-氯卡色林和氯卡色林消旋体各4mg,分别用1mL甲醇溶液溶解后,用正己烷稀释10倍,配制成浓度为400μg/mL的溶液,再取0.4mLR-氯卡色林溶液和0.8mL氯卡色林消旋体溶液混合后得到样品溶液,此时溶液中R-氯卡色林和S-氯卡色林的含量比为2:1,则R-氯卡色林和S-氯卡色林的色谱峰峰面积之比应为2:1;
(2)采用安捷伦1260高效液相色谱仪,以直链淀粉类手性柱为色谱柱,以正己烷、乙醇和二乙胺为正相流动相。控制流动相流速为0.8mL/min,检测波长为270nm,色谱柱柱温箱温度为35℃,进样量为10μL,调节二乙胺在正相流动相中的比例,对上述R/S-氯卡色林样品溶液进行手性分离;
调节二乙胺在正相流动相中的比例,考察二乙胺对分离效果的影响,分为以下几组,按体积百分比计算,即正己烷:乙醇:二乙胺为75%:25%:0%,74.9%:25%:0.1%,74.8%:25%:0.2%,74.7%:25%:0.3%,74.6%:25%:0.4%。
所用的直链淀粉类手性柱为Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm),以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相。
上述的色谱分离结果如下表:
表中t1和对称因子1分别为S-氯卡色林的保留时间和色谱峰对称因子,t2和对称因子2分别为R-氯卡色林的保留时间和色谱峰对称因子,Rs为分离度。
由上表的结构可以看出,本实施例中二乙胺的添加量对色谱峰分离度和对称因子均有影响,流动相中不含二乙胺时,30min内未见出峰,加入少量二乙胺峰形能够得到很大改善,当二乙胺含量从0.3%提高至0.4%,柱效及峰形没有太大改善,结合分离度Rs,故最后所选取的二乙胺最优添加量为0.3%。
当二乙胺的添加量为0.3%时,R/S-氯卡色林的拆分结果见附图1,从图1中可以看出,保留时间为7.69min的为S-氯卡色林,保留时间为9.28min的为R-氯卡色林,两者的分离度Rs为6.36,此时,色谱峰形得到很大改善,两色谱峰达到完全的基线分离。二乙胺主要通过两个方面来减少拖尾,改善峰形,提高分离的选择性,一方面是抑制胺基离子化,另一方面是屏蔽CSP的残余硅烷醇基团。
实施例2:流动相流速对分离效果的影响
(1)取R-氯卡色林和氯卡色林消旋体各4mg,分别用1mL甲醇溶液溶解后,用正己烷稀释10倍,配制成浓度为400μg/mL的溶液,再取0.4mL R-氯卡色林溶液和0.8mL氯卡色林消旋体溶液混合后得到样品溶液,此时溶液中R-氯卡色林和S-氯卡色林的含量比为2:1,则R-氯卡色林和S-氯卡色林的色谱峰峰面积之比应为2:1;
(2)采用安捷伦1260高效液相色谱仪,以直链淀粉类手性柱为色谱柱,以正己烷、乙醇和二乙胺为正相流动相,按体积百分比计算,即正己烷:乙醇:二乙胺为74.7%:25%:0.3%。检测波长为270nm,色谱柱柱温箱温度为35℃,进样量为10μL,调节流动相流速,对上述R/S-氯卡色林样品溶液进行手性分离;
控制流动相流速,考察流动相流速对分离效果的影响,分为以下几组,即流动相流速分别为0.4mL/min,0.6mL/min,0.8mL/min。
所用的直链淀粉类手性柱为Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm),以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相。
上述的色谱分离结果如下表:
表中t1为S-氯卡色林的保留时间,t2为R-氯卡色林的保留时间,Rs为分离度
由上表的结果可以看出,当流速从0.8mL/min减小至0.4mL/min时,Rs只是稍有改变,并未有明显变化。但若继续降低流速,虽可进一步提高分离度,但分析时间也同时增加,峰形也会随之变宽,故最后选取的最优流速为0.8mL/min。此时,两色谱峰分离度较高,出峰时间合适,成本更低。
当控制流速为0.4mL/min,0.6mL/min,0.8mL/min时,R/S-氯卡色林的拆分结果见附图2A、图2B、图2C。图2A的色谱图为当流速为0.8mL/min时R/S-氯卡色林的拆分结果,图2B的色谱图为当流速为0.6mL/min时R/S-氯卡色林的拆分结果,图2C的色谱图为当流速为0.4mL/min时R/S-氯卡色林的拆分结果。
实施例3:正相流动相中醇的种类和比例对分离效果的影响
(1)取R-氯卡色林和氯卡色林消旋体各4mg,分别用1mL甲醇溶液溶解后,用正己烷稀释10倍,配制成浓度为400μg/mL的溶液,再取0.4mLR-氯卡色林溶液和0.8mL氯卡色林消旋体溶液混合后得到样品溶液,此时溶液中R-氯卡色林和S-氯卡色林的含量比为2:1,则R-氯卡色林和S-氯卡色林的色谱峰峰面积之比应为2:1;
(2)采用安捷伦1260高效液相色谱仪,以直链淀粉类手性柱为色谱柱,以正己烷、醇和二乙胺为正相流动相,所述的醇为乙醇或异丙醇;控制流动相流速为0.8mL/min,检测波长为270nm,色谱柱柱温箱温度为35℃,进样量为10μL,调节正相流动相中醇的种类和比例,对上述R/S-氯卡色林样品溶液进行手性分离;
为考虑正相流动相中醇的种类和比例对分离效果的影响,分为以下几组:
上述方法中,正相流动相中的醇为乙醇时,调节乙醇的比例,按体积百分比计算,即正己烷:乙醇:二乙胺为94.7%:5%:0.3%,89.7%:10%:0.3%,84.7%:15%:0.3%,74.7%:25%:0.3%,64.7%:35%:0.3%,54.7%:45%:0.3%;
上述方法中,由正己烷、醇和碱性添加剂组成的正相流动相中,醇为异丙醇时,控制流动相比例,按体积百分比计算,即正己烷:异丙醇:二乙胺为94.7%:5%:0.3%,89.7%:10%:0.3%,84.7%:15%:0.3%,79.7%:20%:0.3%,69.7%:30%:0.3%;59.7%:40%:0.3%。
所用的直链淀粉类手性柱为Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm),以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相。
上述的色谱分离结果如下表:
表中t1为S-氯卡色林的保留时间,t2为R-氯卡色林的保留时间,Rs为分离度
由上表的结果可以看出,当流动相组成为正己烷、乙醇和二乙胺时,流动相比例,按体积百分比计算,即正己烷:乙醇:二乙胺为94.7:5:0.3,84.7:15:0.3,74.7:25:0.3,64.7:35:0.3,54.7:45:0.3(v,v%)变化时,分离度Rs分别为9.00,7.41,6.51,5.80,5.41逐渐减小;同样当流动相组成为正己烷、异丙醇和二乙胺时,按体积百分比计算,即正己烷:异丙醇:二乙胺为94.7:5:0.3,89.7:10:0.3,84.7:15:0.3,79.7:20:0.3,69.7:30:0.3;59.7:40:0.3(v,v%)时,分离度也逐渐减小。且当流动相中的醇分别采用乙醇和异丙醇时,两者虽均可以使R/S-氯卡色林分离,但当流动相组成为正己烷、乙醇和二乙胺时的分离度明显高于当流动相组成为正己烷、异丙醇和二乙胺时,故最后选取的最优流动相组成为正己烷,乙醇和二乙胺。
当流动相组成为正己烷,异丙醇和二乙胺时,流动相比例,按体积百分比计算,即正己烷:异丙醇:二乙胺为84.7:15:0.3(v,v%)时,R/S-氯卡色林的拆分结果见附图3,从图3可以看出S-氯卡色林的保留时间为7.700min,R-氯卡色林的保留时间为8.378min。
当流动相组成为正己烷,乙醇和二乙胺时,流动相比例,按体积百分比计算,以正己烷:乙醇:二乙胺为84.7:15:0.3(v,v%)时的例子,R/S-氯卡色林的拆分结果见附图4,从图4可以看出S-氯卡色林的保留时间为9.090min,R-氯卡色林的保留时间为11.295min。
当乙醇采用25%时,保留时间较短,分离度较好。
实施例4:色谱柱柱温对分离效果的影响
(1)取R-氯卡色林和氯卡色林消旋体各4mg,用1mL甲醇溶液溶解后,用正己烷稀释10倍,配制成浓度为400μg/mL的溶液,再取0.4mLR-氯卡色林溶液和0.8mL氯卡色林消旋体溶液混合后得到样品溶液,此时溶液中R-氯卡色林和S-氯卡色林的含量比为2:1,则R-氯卡色林和S-氯卡色林的色谱峰峰面积之比应为2:1;
(2)采用安捷伦1260高效液相色谱仪,以直链淀粉类手性柱为色谱柱,以正己烷、乙醇和二乙胺为正相流动相,控制流动相比例,按体积百分比计算,即正己烷:乙醇:二乙胺为79.7%:25%:0.3%。控制流动相流速为0.8mL/min,检测波长为270nm,进样量为10μL,调节色谱柱柱温,将上述R/S-氯卡色林样品溶液进行手性分离;
为考虑柱温对分离效果的影响,色谱柱柱温箱温度分为5℃,10℃,15℃,20℃,25℃,30℃,35℃,40℃几组。
所用的直链淀粉类手性柱为Chiralpak IF手性柱(Daicel,250mm×4.6mm,5μm),以直链淀粉-三(3-氯-4-甲基苯基氨基甲酸酯)共价键合在硅胶表面为手性固定相。
上述的色谱分离结果如下表:
表中t1为S-氯卡色林的保留时间,t2为R-氯卡色林的保留时间,Rs为分离度
由上表的结果可以看出,本实施例中不同色谱柱温度对色谱峰分离度有影响,随着色谱柱柱温箱温度的升高,分离度呈下降的趋势,但都能满足完全分离,考虑到分析效率,选择的较短的保留时间,39℃虽然保留时间最短,但对柱子寿命有些影响,因此最后选择的最优色谱柱柱温箱温度为35℃。
最佳色谱柱柱温箱温度为25℃时,R/S-氯卡色林的拆分结果见附图1,从图1可以看出,保留时间为7.69min的为S-氯卡色林,保留时间为9.28min的为R-氯卡色林。
实施例5:不同色谱柱对分离效果的影响
采用不同色谱柱(均为Daicel公司产品)及色谱条件进行分离比较,结果见下表:
经过发明人大量实验,发现色谱柱采用Chiralpak IF手性柱才能将R/S-氯卡色林较好地分离。
- 还没有人留言评论。精彩留言会获得点赞!