一个植物谷蛋白转运储藏相关蛋白GPA4及其编码基因与应用的制作方法
本发明属于基因工程领域,涉及一个植物谷蛋白转运贮藏相关蛋白GPA4及其编码基因与应用。
背景技术:
高等植物种子中往往积累大量的储藏性物质,作为种子萌发的营养来源,同时也是人类和畜牧业重要的蛋白质来源。水稻(Oryza sativa L.)是世界上最重要的粮食作物之一,全球超过50%人口以稻米为主食。水稻种子在胚乳中积累大量的储藏蛋白,它是稻米中仅次于淀粉的第二大物质,约占籽粒干重的8-10%,其中谷蛋白可占60-80%,醇溶蛋白约占10-20%,球蛋白约为5-10%。稻米中蛋白的含量和组成直接影响稻米的外观、营养和食味品质等特性。因此,深入研究水稻储藏蛋白合成、加工、分选、积累的分子机制对培育满足不同人群需求的蛋白含量各异的水稻新品种具有重要意义。
水稻种子中的谷蛋白贮存在不规则的II型在蛋白体中(Protein Body II;水稻中PBII实质是蛋白质储藏液泡PSV,Protein Storage Vacuoles),可以被人体消化吸收,且谷蛋白富含人体必需氨基酸,是稻米蛋白品质改良的首选目标;醇溶蛋白因储存在致密的蛋白体PBI(Protein Body I)中而不能被消化。水稻谷蛋白最初以57-kDa前体的形式在内质网ER(Endoplasmic Reticulum)表面的多聚核糖体(Polysomes)上合成,在信号肽的引导下边合成边进入内质网囊腔,谷蛋白前体在内质网囊腔中剪切掉信号肽(Signal Peptide,SP),并接受Bip(binding protein)、PDI(protein disulfied isomerase)等分子伴侣的辅助折叠形成正确的构象后,谷蛋白通过由内质网衍生而成的类前体蛋白积累区室(precursor accumulating compartment like,PAC)直接进入PBII中;或被转运至Golgi体并进一步进行加工修饰,通过在反面高尔基体管网结构(Trans-Golgi networks,TGN)形成的致密囊泡(Dense vesicles,DV)被分泌到细胞膜外,但由于谷蛋白带有液泡分选信号(Vacuolar Sorting Signal,VSS),VSS引导DV进入PBII,进入PBII的谷蛋白前体在液泡加工酶OsVPE1(Vacuolar Processing Enzyme)的作用下水解为40-kDa的酸性亚基和20-kDa的碱性亚基。如果上述途径的任一过程存在缺陷,谷蛋白将会以57-kDa前体的形式积累在胚乳细胞中,导致PBII的充实度降低,从而影响稻米中可被人体吸收的成熟谷蛋白的含量、积累和分布。
基于上述策略,我们通过大规模的突变体筛选,获得了一份新的谷蛋白57H突变体,命名为gpa4(glutelin precursor accumulation 4)(gpa4-1为9311辐射诱变突变体(Indica遗传背景),gpa4-2位Kinmaze MNU处理突变体(Japonica)))。利用图位克隆的方法获得了GPA4基因。GPA4 CDS(coding sequence)长423bp,编码一个包含四个跨膜区的膜蛋白,酵母中GPA4的同源蛋白Got1p的缺失突变体没有可见的表型,认为酵母Got1是一个非必需基因,而Got1和sft2的双突变体则会影响酵母细胞中蛋白质在ER和Golgi中的运输。动物中也有GPA4同源蛋白的报道,如human Got1A和Got1B,然而它们的功能均未知。
技术实现要素:
:
本发明的目的是提供一个植物谷蛋白转运储藏相关蛋白及其编码基因与应用。
本发明提供的谷蛋白转运储藏相关蛋白(GPA4),来源于稻属水稻(Oryza sativa var.9311或Kinmaze),是如下(a)或(b)的蛋白质:
(a)由SEQ ID NO.1所示的氨基酸序列组成的蛋白质;
(b)将EQ ID NO.1的氨基酸序列经过一个或几个氨基酸残基的取代和/或缺失和/或添加且与植物谷蛋白转运储藏相关的由SEQ ID NO.1衍生的蛋白质。
SEQ ID NO.1由140个氨基酸残基组成,为了使(a)中的GPA4便于纯化,可在由SEQ ID NO.1所示的氨基酸序列组成的蛋白质的氨基末端或羧基末端连接上如表1所示的标签。
表1融合标签的序列
上述(b)中的GPA4可人工合成,也可先合成其编码基因,再进行生物表达得到。
上述(b)中的GPA4的编码基因可通过将SEQ ID NO.2所示的DNA序列中缺失一个或几个氨基酸残基的密码子,和/或进行一个或几个碱基对的错义突变,和/或在其5′端和/或3′端连上
编码上述植物谷蛋白运输储藏相关蛋白的基因GPA4也属于本发明的保护范围。
所述基因可为如下1)或2)或3)或4)的DNA分子:
1)SEQ ID NO.2所示的DNA分子;
2)SEQ ID NO.3所示的DNA分子;
3)在严格条件下与1)或2)限定的DNA序列杂交且编码所述蛋白的DNA分子;
4)与1)或2)或3)限定的DNA序列具有90%以上同源性,且编码植物谷蛋白转运储藏相关蛋白的DNA分子。
所述严格条件可为在0.1×SSPE(或0.1×SSC),0.1%SDS的溶液中,在65℃下杂交并洗膜。
SEQ ID NO.2由423个核苷酸组成,为基因GPA4的编码区CDS。
含有以上任一所述基因的重组表达载体。
可用现有的植物表达载体构建含有所述基因的重组表达载体。
所述植物表达载体包括双元农杆菌载体和可用于植物微弹轰击的载体等。所述植物表达载体还可包含外源基因的3’端非翻译区域,即包含聚腺苷酸信号(poly A)和任何其它参与mRNA加工或基因表达的DNA片段。所述聚腺苷酸信号可引导聚腺苷酸加入到mRNA前体的3’端,如农杆菌冠瘿瘤诱导(Ti)质粒基因(如胭脂合成酶Nos基因)、植物基因(如大豆贮存蛋白基因)3’端转录的非翻译区均具有类似功能。
使用所述基因构建重组植物表达载体时,在其转录起始核苷酸前可加上任何一种增强型启动子或组成型启动子,如花椰菜花叶病毒(CAMV)35S启动子、玉米的泛素启动子(Ubiquitin),它们可单独使用或与其它的植物启动子结合使用;此外,使用本发明的基因构建植物表达载体时,还可使用增强子,包括翻译增强子或转录增强子,这些增强子区域可以是ATG起始密码子或邻接区域起始密码子等,但必需与编码序列的阅读框相同,以保证整个序列的正确翻译。所述翻译控制信号和起始密码子的来源是广泛的,可以是天然的,也可以是合成的。翻译起始区域可以来自转录起始区域或结构基因。
为了便于对转基因植物细胞或植物进行鉴定及筛选,可对所用植物表达载体进行加工,如加入可在植物中表达的编码可产生颜色变化的酶或发光化合物的基因(GUS基因、绿色荧光蛋白基因等)、具有抗性的抗生素标记物(庆大霉素标记物、卡那霉素标记物等)或是抗化学试剂标记基因(如抗除草剂基因)等。
所述重组表达载体优选为在pCUBi1390的酶切位点BamHI之间插入所述基因GPA4得到的重组质粒,命名为pCUBi1390-GPA4。
含有以上任一所述基因(GPA4)的表达盒、转基因细胞系及重组菌。扩增所述基因(GPA4)全长或任一片段的引物对也属于本发明的保护范围。
本发明的另一个目的是提供一种培育成熟谷蛋白含量增加的植物的方法。
本发明提供的培育成熟谷蛋白含量增加的植物的方法,是将所述基因导入成熟谷蛋白含量降低的植物中,得到成熟谷蛋白含量正常的转基因植物;所述谷蛋白含量降低植物为籽粒中成熟谷蛋白含量显著低于正常型的植物;所述谷蛋白含量正常的转基因植物为籽粒中成熟谷蛋白的含量与正常型相当的转基因植物。具体来说,所述基因通过所述重组表达载体导入成熟谷蛋白含量降低的植物中;所述成熟谷蛋白含量降低的植物可为gpa4(gpa4-1和gpa4-2)。
所述蛋白、所述基因、所述重组表达载体、表达盒、转基因细胞系或重组菌或所述方法均可应用于水稻育种。
利用任何一种可以引导外源基因在植物中表达的载体,将编码所述蛋白的基因导入植物细胞,可获转基因细胞系及转基因植株。携带有所述基因的表达载体可通过使用Ti质粒、Ri质粒、植物病毒载体、直接DNA转化、显微注射、电导、农杆菌介导等常规生物学方法转化植物细胞或组织,并将转化的植物组织培育成植株。被转化的植物宿主既可以是单子叶植物,也可以是双子叶植物,如:烟草、百脉根、拟南芥、水稻、小麦、玉米、黄瓜、番茄、杨树、草坪草、苜宿等。
有益效果:
本发明首次发现、定位并克隆得到一个新的植物谷蛋白运输储藏相关蛋白的基因GPA4。本发明的植物谷蛋白转运储藏相关蛋白影响植物的囊泡运输过程。抑制该蛋白编码基因的表达可导致植物种子中谷蛋白的囊泡运输过程的障碍,并影响其剪切成熟,从而可以培育囊泡运输变异的转基因植物和植物成熟谷蛋白含量降低的转基因植物。将所述蛋白的编码基因导入成熟谷蛋白含量降低的植物中,可以培育成熟谷蛋白含量正常的植物。所述蛋白及其编码基因可以应用于植物遗传改良。
附图说明
图1为GPA4基因在水稻第3染色体上的精细定位;
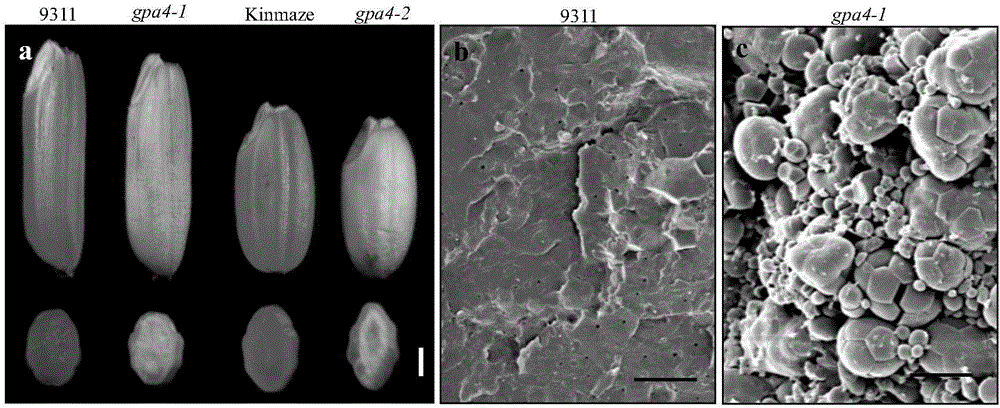
图2为野生型和突变体的成熟籽粒的扫描电镜观察。
图3为野生型和突变体成熟种子中总蛋白的SDS-PAGE分析。
图4为野生型9311和突变体gpa4-1发育中期胚乳细胞形态比较。
图5为野生型9311和突变体gpa4-1发育胚乳细胞中贮藏蛋白免疫荧光分析。
图6为野生型9311和突变体gpa4-1发育胚乳细胞的透射电镜观察。
图7为转pCUbi1390-GPA4的T0代植株的透明籽粒外观。
图8为转pCUbi1390-GPA4的T0代植株透明籽粒的SDS-PAGE电泳图谱。
图9为转pCUbi1390-GPA4的T0代植株籽粒的透射电镜观察。
具体实施方式
以下的实施例便于更好地理解本发明,但并不限定本发明。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为购自常规生化试剂商店。
实施例1、植物谷蛋白分选相关蛋白及其编码基因的发现
一、水稻谷蛋白分选突变体gpa4表型分析及其遗传分析
在9311的60Co辐射突变体库中筛选出籽粒粉质突变体gpa4-1,在Kinmaze MNU处理突变体库中筛选出粉质突变体gpa4-2,两个突变体为同一个基因的等位变异。野生型相比比较,gpa4的主要特征是籽粒粉质,不透明(见图2)。扫描电镜分析证实,gpa4-1中淀粉颗粒松散(见图2),可能是导致胚乳不透明的主要原因。此外,突变体成熟种子的SDS-PAGE分析表明基因的突变导致为57-kDa的谷蛋白前体剧增,相应的成熟的酸性亚基和碱性亚基含量降低,与此同时伴随球蛋白含量下降,醇溶蛋白未表现出明显差异(见图3)。
水稻胚乳中存在两类蛋白体,分别为蛋白体I(Protein Body I,PBI)和蛋白体II(PBII)。前者贮存有醇溶蛋白组成,后者由谷蛋白和球蛋白构成。发育胚乳的半薄切片经考马斯亮蓝染色,光学显微镜观察显示(见图4):野生型9311和gpa4-1胚乳细胞内部蛋白体的大小存在显著差异,突变体中存在大量的小的蛋白体结构,图4的结果表明突变体gpa4-1中蛋白体变小。
制备发育胚乳半薄切片,并采用免疫荧光标记发现,野生型种PBII较大,而突变体中,谷蛋白和醇溶蛋白并未相互交联,而是分开贮藏。在突变体中,我们发现了一种新型结构,该结构带有一个谷蛋白的核,四周被醇溶蛋白的聚集体包围(见图5,箭头所示)。对发育胚乳的透射电镜观察显示突变体中存在大量的异常结构,该异常结构有一个电子密度致密的核,四周被电子密度较低的蛋白聚集体包围。同时该结构被内质网膜包裹,显示该结构是内质网衍生的结构,暗示储藏蛋白出ER存在缺陷(图6a-e)。在种子发育的中后期,突变体中还大量出现内质网衍生的另一种结构,该结构贮存有醇溶蛋白(图6f)。
综上所述,在突变体gpa4中由于谷蛋白的ER输出存在缺陷,导致谷蛋白滞留下ER囊腔中,逐步形成第一种特殊的结构(谷蛋白核+醇溶蛋白外周),由于蛋白的大量滞留,导致ER正常的功能受到损害,PBI的形成受到影响,从而形成大量的未成熟的PBI。
突变体gpa4-1与其野生型9311杂交,得到杂交组合gpa4-1/9311,对F2分离群体中239粒种子调查结果显示,透明种子与不透明粉质种子的分离比符合3:1,暗示突变基因由一对隐性基因控制,且籽粒不透明性状与成熟谷蛋含量降低性状共分离。
二、突变基因的定位
1、突变基因的初定位
用突变体gpa4-1(Indica)和日本晴(Japonica)杂交,在gpa4-1/Nipponbare的F2分离群体中随机选取籽粒粉质且表现为成熟谷蛋白含量降低的种子,包括两个亲本的种子发芽后,提取DNA。在全基因组范围内分析gpa4-1和日本晴之间的多态性,之后每间隔10-15cM挑选一对在两个亲本间有多态的引物。两个亲本DNA加10个隐性极端个体的DNA,进行全基因组的连锁分析,最后将谷蛋白分选关键基GPA4(Glutelin Precursor Accumulation 4)定位在第3染色体SSR标记RM175和RM7576之间12.3cM的区域。
上述SSR标记分析的方法如下所述:
(1)提取上述选取单株的总DNA作为模板,具体方法如下:
①取0.2克左右的水稻幼嫩叶片,置于2mL离心管中,管中放置一粒钢珠,把装好样品的离心管在液氮中冷冻5min,置于2000型GENO/GRINDER仪器上粉碎样品1min。
②加入660μl提取液(含100mM Tris-Hcl(pH 8.0),20mM EDTA(pH 8.0),1.4M NaCl,0.2g/mL CTAB的溶液),漩涡器上剧烈涡旋混匀,冰浴30min。
③加入40μL 20%SDS,65℃温浴10min,每隔两分钟轻轻上下颠倒混匀。
④加入100μL 5M NaCl,温和混匀。
⑤加入100μL 10×CTAB,65℃温浴10min,间断轻轻上下颠倒混匀。
⑥加入900μL氯仿,充分混匀,12000rpm离心3min。
⑦转移上清液至1.5mL Eppendorf管中,加入600μL异丙醇,混匀,12000rpm离心5min。
⑧弃上清液,沉淀用70%(体积百分含量)乙醇漂洗一次,室温凉干。
⑨加入100μL 1×TE溶解DNA。
⑩取2μL电泳检测DNA质量,并用DU800分光光度仪测定浓度(Bechman Instrument Inc.U.S.A)。
(2)将上述提取的DNA稀释成约20ng/μL,作为模板进行PCR扩增;
PCR反应体系(10μL):DNA(20ng/μL)1μL,上游引物(2pmol/μL)1μL,下游引物(2pmol/ul)1μL,10×Buffer(MgCl2free)1μL,dNTP(10mM)0.2μL,MgCl2(25mM)0.6μL,rTaq(5U/μL)0.1μL,ddH2O 5.1μL,共10μL。
PCR反应程序:94.0℃变性5min;94.0℃变性30S、55℃退火30S、72℃延伸1min,35个循环;72℃延伸3min;10℃保存。PCR反应在Applied Systems Veriti 96孔热循环仪中进行。
(3)SSR标记的PCR产物检测
扩增产物用8%非变性聚丙烯酰胺凝胶电泳分析。以50bp的DNA Ladder为对照比较扩增产物的分子量大小,银染显色。
2、突变基因精细定位
根据初步定位的结果,在突变位点所在区域间隔一定区段自行开发SSR标记,以便在该染色体的相关区段筛选更多标记进一步定位突变体位点。从gpa4-1/Nipponbare杂交组合获得的F2分离群体中挑出确认为突变表型的F2种子,用于突变位点的精细定位。利用公共图谱上的分子标记和基于水稻基因组序列数据自行开发的SSR、CAPS、dCAPS、InDel等分子标记对突变位点进行了精细定位,并根据定位结果初步确定突变位点,具体方法如下:
(1)SSR标记开发
将公共图谱的SSR标记与水稻基因组序列进行整合,下载突变位点附近的BAC/PAC克隆序列。用SSRHunter(李强等,遗传,2005,27(5):808-810)或SSRIT软件搜索克隆中潜在的SSR序列(重复次数≥10);将这些SSR及其邻近400~500bp的序列在NCBI通过BLAST程序在线与相应的籼稻序列进行比较,如果两者的SSR重复次数有差异,初步推断该SSR引物的PCR产物在籼、粳间存在多态性;再利用Primer Premier 5.0软件设计SSR引物,并由南京金斯瑞合成。将自行设计的SSR成对引物等比例混合,检测其在gpa4-1和日本晴之间的多态性,表现多态者用作精细定位GPA4-1基因的分子标记。用于精细定位的分子标记见表2。
表2 GPA4基因精细定位所用的引物
根据F2群体中胚乳粉质,成熟谷蛋白含量降低单株的分子数据和表型数据,按照Zhang等报道的“隐性极端个体基因作图”方法,最终把GPA4基因精细定位在BAC克隆OSJNBa0014O06和OSJNBa0021B21重叠的50kb的区域内,位于SSR标记G11和G15之间(图1)。对候选区段基因组测序显示,在gpa4-1中,基因Os03g0209400缺失了256bp,导致该基因的cDNA缺失了108bp,蛋白只正确地翻译了最初的10个氨基酸残基;而gpa4-2中发生单个碱基的变异,导致47位甘氨酸突变为天冬氨酸。因此,gpa4-1可能是Os03g0209400的功能丧失突变体,而gpa4-2是功能部分丧失突变体。
(2)突变基因的获得
根据定位的位点设计引物,序列如下所述:
primer1:
5'GAAAACGCCGCTACGCTACCAC 3'(SEQ ID NO.4)
primer2:
5'GCTCAAGCATAAGCCGACAAGG 3'(SEQ ID NO.5)
以primer1和primer2为引物,以水稻Kinmaze的cDNA为模板,进行PCR扩增获得目的基因。该对引物位于SEQ ID NO.2上游149bp和下游359bp,扩增产物为目的基因的5’UTR区,CDS区(ATG到TGA,ATG红色字体标出,TGA蓝色字体标出)和3’UTR区。扩增反应在Applied Biosystems Veriti 96孔PCR仪上进行:94℃3min;94℃℃30sec,55℃℃30sec,72℃℃1min,32个循环;72℃℃3min。将PCR产物回收纯化后克隆到载体pEASY(北京全式金公司),转化大肠杆菌DH5α感受态细胞(北京Tiangen公司),挑选阳性克隆后,进行测序。
序列测定结果表明,PCR反应获得的片段具有序列表中SEQ ID NO.2所示的核苷酸序列,编码140个氨基酸残基组成的蛋白质(从ATG到TGA)(见序列表的SEQ ID NO.1)。将SEQ IDNO.1所示的蛋白命名为GPA4,将SEQ ID NO.1所示的蛋白的编码基因命名GPA4。
实施例2、转基因植物的获得和鉴定
一、重组表达载体构建
以水稻Kinmaze的cDNA为模板,进行PCR扩增获得GPA4基因的编码区,PCR引物序列如下:
primer3:(下划线为BamHI酶切位点)
5'CTGCAGGTCGACGGATCCATGGTTTCCTTCGAGATG 3'(SEQ ID NO.6)
primer4:(下划线为BamHI酶切位点)
5'TAGAATTCCCGGGGATCCCTACACTGGAACCCGTTT 3'(SEQ ID NO.7)
上述扩增产物包含了该基因的编码区,将PCR产物回收纯化。采用Infusion重组试剂盒(Clontech)将PCR产物克隆到载体pCUbi1390中,构建成pCUbi1390-GPA4。重组反应体系(2.5μL):PCR产物0.5μL(50-100ng),pCUbi1390载体1.0μL(30-50ng),5×Infusion buffer 2.0μL,Infusion enzyme mix 0.5μL。短暂离心后将混合体系50℃℃水浴15min,反应结束后迅速取出插入冰上,将2.5μL反应体系用热激法转化大肠杆菌DH5α感受态细胞(北京Tiangen公司;CB101)。将全部转化细胞均匀涂布在含50mg/L卡那霉素的LB固体培养基上。
37℃℃培养16h后,挑取克隆阳性克隆,进行测序。测序结果表明,得到了含有SEQ ID NO.2所示基因的重组表达载体,将含有GPA4 cDNA序列的pCUbi1390命名为pCUbi1390-GPA4,GPA4基因插入在多克隆位点BamHI之间。
二、重组农杆菌的获得
用冻融法将pCUbi1390-GPA4转化农杆菌EHA105菌株(购自美国英骏公司),得到重组菌株,提取质粒进行PCR及酶切鉴定。将PCR及酶切鉴定正确的重组菌株命名为EH-pCUbi1390-GPA4。
三、转基因植物的获得
分别将EH-pCUbi1390-GPA4转化突变体gpa4-2(gpa4-1为Indica背景,转化难度较大),具体方法为:
(1)28℃培养EH-pCUbi1390-GPA4培养16h,收集菌体,并稀释到含有100μmol/L乙酰丁香酮的N6液体培养基(Sigma公司,C1416)中至浓度为OD600≈0.6,获得菌液;
(2)将培养至一个月的gpa4-2水稻成熟胚胚性愈伤组织与步骤(1)的菌液混合侵染30min,滤纸吸干菌液后转入共培养培养基(N6固体共培养培养基,Sigma公司)中,24℃共培养3天;
(3)将步骤(2)的愈伤接种在含有50mg/L潮霉素(Roche公司)的N6固体筛选培养基上第一次筛选(15天);
(4)挑取健康愈伤转入含有100mg/L潮霉素的N6固体筛选培养基上第二次筛选,每15天继代一次;
(5)挑取健康愈伤转入含有50mg/L潮霉素的N6固体筛选培养基上第三次筛选,每15天继代一次;
(6)挑取抗性愈伤转入分化培养基上分化;得到分化成苗的T0代阳性植株。
四、转基因植株的鉴定
1、PCR分子鉴定
将步骤三获得的T0代植株提取基因组DNA,以基因组DNA为模板,利用Primer5和Primer6为引物对进行扩增,序列如下:
Primer5:
5'GATGAATGACCTCAAGAAGATTGG 3'(SEQ ID NO.8)
Primer6:
5'AACCCGTTTTCCTCTGTAGCG 3'(SEQ ID NO.9))
PCR反应体系:DNA(20ng/μL)2μL,Primer5(10pmol/μL)2μL,Primer6(10pmol/μL)2μL,10×Buffer(MgCl2free)2μL,dNTP(10mM)0.4μL,MgCl2(25mM)1.2μL,rTaq(5U/μL)0.4μL,ddH2O 10μL,总体积20μL。扩增反应在Applied Biosystems Veriti 96孔PCR仪上进行:94℃3min;94℃30sec,55℃45sec,72℃1min,32个循环;72℃3min。用试剂盒(北京Tiangen公司)纯化回收PCR产物。PCR产物用1%的琼脂糖电泳检测。
2、表型鉴定
分别将T0代转pCUbi1390-GPA4的植株,Kinmaze和gpa4-2种植在南京农业大学牌楼网室内。转基因后代家系中出现了透明的籽粒(透明:不透明=3:1)(图7),提取种子总蛋白进行SDS-PAGE检测透明种子与Kinmaze表现一样(图8)。同时制备发育胚乳的超薄切片观察发现,互补家系的胚乳细胞中异常结构消失,与野生型细胞无差异(图9)。由此验证了转基因前的不透明粉质和谷蛋白前体增加性状是由GPA4基因控制的,即该GPA4基因为谷蛋白分选关键基因。并且,将pCUbi1390-GPA4转化的水稻gpa4-2突变体,可以使其成熟谷蛋白含量上升至正常水平。
- 还没有人留言评论。精彩留言会获得点赞!