一种终止双等位基因转录的方法与流程
本发明涉及基因工程领域,具体涉及一种终止双等位基因转录的方法。
背景技术:
基因表达的缺失在研究基因功能中起到十分重要的作用。常规的造成基因表达缺失的策略是进行RNA干扰,然而由于RNA的高脱靶率以及不完全的敲除,很大程度上限制了该方法的广泛应用。利用基因编辑工具介导的编码基因的移码突变,或者针对整个基因或启动子的大片段删除可以有效地实现基因敲除或者敲低的目的,但是大片段的删除可能导致其他基因相关功能原件的丢失,造成基因功能的误判。此外,为了在基因敲除中获得双等位基因敲除细胞株,需要大量时间进行单克隆细胞株筛选工作。
CRISPR/CAS系统是一种细菌和古细菌用于外源遗传物质的降解的适应性免疫防御机制,也可用于包括哺乳动物细胞在内的各种系统的基因编辑中。使用CRISPR/Cas9敲除编码基因时,可以通过目的基因碱基的插入和缺失进行敲除。由于二倍体细胞的大部分基因含有两个等位基因,在进行了CRISPR/Cas9质粒转染后,在部分细胞中,目的基因的敲除会发生在两个等位基因上(双等位基因敲除),然而有些细胞中,只敲除了一个等位基因(单等位基因敲除),也就是说,CRISPR/Cas9转染的细胞种群包含了双等位基因和单等位基因敲除的细胞。因此,为了进一步确认哪些是双等位基因敲除的细胞克隆,可能需要额外的转染和筛选,并通过Western Blot实验进行检测,以产生纯合子的基因敲除细胞群。同样,CRISPR/Cas9介导的同源重组在二倍体细胞中也存在相似的问题,即在部分细胞中出现单等位基因的同源重组,而在少部分细胞中为两个等位基因的同源重组。其中由于两个等位基因均同源重组的细胞数量过少,给筛选工作带来更大困难。
中国专利申请CN201010592737.9公开了一种等位基因双敲除打靶载体系统,该系统由pGT-V1和pGT-V2两个互补载体组成,每个载体都包含两个同向的LoxP元件,其间含有两个正筛选标记基因,外侧含有一个负筛选标记基因,即pGT-V1带有正筛选标记新霉素磷酸转移酶基因和绿色荧光蛋白基因,pGT-V2带有正筛选标记潮霉素基因和红色荧光蛋白基因,二者都含有负筛选标记单纯疱疹病毒胸苷激酶基因。该发明申请的技术方案中,经过药物和荧光双筛选标记的选择,一次性实现对靶基因两个等位基因的遗传修饰或敲除,从而提高基因打靶的效率,缩短实验时间。尽管该技术方案理论上可行,但是在实际操作中可能会存在众多问题,例如:1)在哺乳动物胚胎细胞中,同源重组的发生率极低,因此,如果依赖细胞自身的同源重组,即使能够实现,等位基因的打靶也是非常困难的,如果同时实现同一细胞双等位基因同时打靶则更为困难;相对于传统的胚胎细胞基因打靶,终末分化的细胞由于自然发生的同重组率低,导致依赖常规同源重组的基因敲除更加困难;这也是该专利中并未能给出细胞打靶的实例的原因,同样该方案没有实际的应用价值;2)该技术方案中未能给出细胞打靶的实例,即使该系统使用同源重组的方法进行,也只能是依照传统的基因重组,数千bp的同源重组臂给同源重组载体的构建带来了很大的难度;3)该系统仅针对编码基因的打靶,并未涉及非编码基因的敲除。
技术实现要素:
本发明的目的是建立了一种终止双等位基因转录的方法,该方法是一种CRISPR/Cas9介导的、在双等位基因内同时整合含有双筛选标签的转录终止信号以高效准确地终止基因转录、并且高效建立纯合子的双等位基因敲除细胞株的方法。其主要原理是:首先应用CRISPR/Cas技术,在细胞内靶向基因区域诱导较高频率的同源重组事件的发生,极大提高同源重组的效率(无论是胚胎细胞中或终末分化细胞中);其次,使用在目的基因中通过同源重组插入PolyA信号,以达到终止基因转录敲除基因的目的;最后,建立含有双筛选标签(嘌呤霉素抗性基因PuroR和新霉素抗性基因NeoR)的PolyA同源重组载体,所述的同源重组载体在改变上游与下游的同源臂后,在CRISPR/Cas9介导的双链断裂(Double strand break,DSB)修复作用下,能够高效地在两条等位基因上通过同源重组插入含有两个不同筛选标签的PolyA序列,随后使用双抗生素进行筛选。依靠以上三个优势,实现高效定点插入的同时,快速获得双等位基因同时敲除的细胞株的目的。
为了实现上述目的,本发明的技术方案如下:
本发明提供了一种用于终止双等位基因转录的同源重组载体,其特征在于,所述同源重组载体的每个同源重组载体包括插入片段,所述的插入片段包括剪切受体(splice adopter,SA)、自我剪切2A肽(Self-cleaving 2A peptide)(如:T2A、P2A、E2A或者F2A等)、转录终止信号(Termination signal)片段和抗生素筛选基因,其中,所述每对同源重组载体之间具有两种不同的抗生素筛选基因,以方便筛选,所述转录终止信号片段位于抗生素筛选基因下游。
可选地,所述的抗生素筛选基因为新霉素抗性基因(NeoR)或者嘌呤霉素抗性基因(PuroR),其核苷酸序列分别为SEQ ID NO.1和SEQ ID NO.2。抗性基因的选择并不仅限于这两个基因,可以使任意两种无相互影响的抗性基因。
可选地,所述的转录终止信号为SV40PolyA(Simian vacuolating virus 40PolyA)、β-globin teminator或者BGH PolyA(bovine growth hormone PolyA),其核苷酸序列分别为SEQ ID NO.3、SEQ ID NO.4和SEQ ID NO.5;并且不仅限于该三种终止信号,其他的终止信号也可以,只要满足a)终止效率高;b)片段较小容易整合的特点。优选地,使用BGH PolyA作为转录终止信号,其优点如下:1)BGH PolyA研究相对明确,应用范围广,且片段相对较小有利于同源重组的插入;2)BGH PolyA序列终止作用是用方向性的,其正向的终止活性较强,而反向的序列终止活性相对较弱;该转录终止信号特别适合针对反义RNA的研究,仅仅沉默DNA正义链的转录,而对反义链影响较弱,反之亦然。
优选地,所述的同源重组载体的插入片段包括含有嘌呤霉素抗性基因(PuroR)的SA-T2A-PuroR-BGH PolyA和含有新霉素抗性基因(NeoR)的SA-T2A-NeoR-BGH PolyA;前者的核苷酸序列如SEQ ID NO.6所示,该序列用于在靶向基因的特异位点同源重组,并在靶向位置插入SA-T2A-PuroR-BGH PolyA序列;后者的核苷酸序列如SEQ ID NO.7所示,该序列用于在靶向基因的特异位点同源重组,在靶向位置插入SA-T2A-NeoR-BGH PolyA序列。
本发明还提供了一种终止双等位基因转录的方法,其特征在于,包括以下步骤:
1)构建如上所述的分别含有两种不同抗生素筛选基因(可选地,嘌呤霉素抗性基因(PuroR)或者新霉素抗性基因(NeoR))的SA-T2A-抗生素筛选基因-PolyA DNA片段;其中,在上述终止信号中优选BGH PolyA,构建SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA DNA片段;
2)根据不同靶向基因设计不同的gRNA,构建含有靶向基因的Cas9/gRNA表达载体,例如,实施例中构建的gRNA载体靶向AAVS1基因;
3)在靶向基因中插入步骤1)中所获得的分别含有两种不同抗生素筛选基因的SA-T2A-抗生素筛选基因-PolyA DNA片段,构建含有靶向基因的同源重组载体;可选地,当步骤2)中选用的含有靶向基因的Cas9/gRNA表达载体的靶向目的基因组为AAVS1基因位点,在AAVS1靶向位点上下游各取800bp分别作为左侧同源臂和右侧同源臂,与步骤1)中获得的SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA片段连接,构建AAVS1-SA-T2A-PuroR-BGH PolyA和AAVS1-SA-T2A-NeoR-BGH PolyA同源重组载体;
4)导入细胞:将步骤3)中所获得的含有靶向基因的同源重组载体,以及步骤2)中所获得的含有靶向基因的Cas9/gRNA表达载体根据实验需要共转染不同类型的细胞,如:HEK293细胞;
5)使用步骤1)中所使用的抗生素基因的相关抗生素筛选至少2周,获取双等位基因被敲除的多克隆细胞株。
本发明的技术方案达到的有益效果是:
1)使用CRISPR/Cas9系统介导,将转录终止信号(如:PolyA)作为元件插入靶向基因中,且所述PolyA信号片段位于抗生素筛选基因下游,并且与抗性基因共同整合至靶基因启动子下游达到终止靶向基因转录,最终实现基因敲除的目的。所述PolyA信号可以a)保证抗性基因的正常转录与翻译,同时终止了抗性基因下游的转录;b)PolyA信号插入目的基因中,不但可以终止抗性基因下游的转录,同时也终止了目的基因在PolyA下游部分的转录(两者同时进行的)。与现有的基因敲除方法相比,本发明提供的基因敲除方法不仅快速、精准和成功率高,而且不会导致其他基因相关功能元件的丢失,确保了基因功能鉴定更准确。
2)由于CRISPR/Cas9靶向DNA序列高效的特异性和同源重组,本发明所述的方法能够高效地对靶向基因进行修饰,并且使得双等位基因终止基因转录;同时由于同源重组的特异性也进一步提高了获得双等位基因敲除的准确性。
3)双抗生素筛选的方法中,采用双正筛选的机制,保证可以快速准确地筛选出同源重组载体被稳定地插入、并且顺利表达的基因组,以确保高效筛选出双等位基因敲除的细胞株,提高基因修饰细胞的筛选效率,以高效地产生纯合子的基因敲除细胞株。
4)本发明所述的方法,适用于细胞系的基因转录终止,进而确定基因功能。本发明提供的含有PuroR-BGH PolyA和NeoR-BGH PolyA元件的同源重组载体,本专业人员可以根据不同的不同靶位设计相应的同源重组序列,以达到适用于不同物种细胞系,进行基因转录终止基因表达敲除的目的,为基因功能缺失的研究提供有效方法。
附图说明
图1是筛选不同的终止信号。将终止信号插入pIRES-EGFP多克隆位点,并转染细胞。
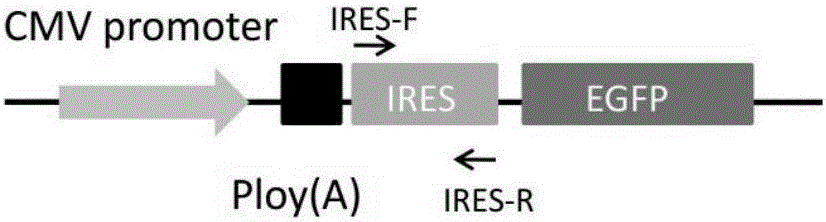
图2是检测IRES引物靶向示意图。
图3是IRES序列RNA的转录水平。
图4是通过重叠PCR扩增后获得的SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA片段示意图。
图5是实施例1中构建的AAVS1同源重组载体的结构示意图。扩增靶向AAVS1位点的上下游共1600bp DNA,并克隆至T载体中。在通过反向PCR,在AAVS1靶向位点将该载体线性化后,分别与SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA进行连接。
图6是含有双筛选标签载体分别与两条等位基因同源重组示意图。
图7是在同源重组区域上下游分别设计引物,用于鉴定SA-T2A-PuroR-BGH PolyA或SA-T2A-NeoR-BGH PolyA插入的细胞株。
图8是使用同源重组区域上下游引物引物鉴定等位基因均插入SA-T2A-PuroR-BGH PolyA或SA-T2A-NeoR-BGH PolyA的细胞株。其中,1.以含有多克隆基因组与未转染基因组的DNA作为模板,扩增显示3条带,即:野生型1796bp、PolyA插入序列扩增条带3310bp和3654bp;2.以使用本发明的方法两条等位基因均插入PolyA的单克隆细胞系基因组DNA为模板,仅含有PolyA插入的两条带分别为3310bp和3654bp。
图9是检测PPP1R12C基因的转录情况的RT-qPCR检测结果。使用同源重组区域上下游引物鉴定后,随机挑选取双等位基因敲除的三个克隆,分别命名为clone1、clone2和clone3,检测该克隆的PPP1R12C mRNA水平。以GAPDH作为内参。
具体实施方式
为了阐明本发明的技术方案及技术目的,下面结合附图及具体实施方式对本发明做进一步的介绍。下述实施例中的方法,如无特别说明,均为常规方法。
实施例1
本实施例1提供了如何将本发明方法应用于针对PPP1R12C基因进行的靶向敲除。其中,选择AAVS1(位于PPP1R12C第一个内含子)为靶向区域,构建含有双抗性基因的同源重组载体AAVS1-PuroR-BGH PolyA和AAVS1-NeoR-BGH PolyA,利用CRISPR/Cas9介导的DNA双链断裂,将其通过同源重组插入PPP1R12C第一个内含子(AAVS1基因区域),终止PPP1R12C基因的表达,最后使用双抗性基因进行高效筛选获得双等位基因同时敲除的细胞株。
根据不同靶向基因,可以通过设计并修改为所需靶向基因相应的同源臂序列以及(CRISPR/Cas所靶向的基因的)gRNA序列,从而将本方法应用于包括人在内的不同物种细胞株基因转录终止。
本实施例1具体包括以下步骤:
1.构建含有双筛选标签的PolyA DNA片段:SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA:
1.1合成SA-T2A序列:
根据参考文献Hockemeyer et al.,2009(Hockemeyer et al Nat Biotechnol.2009Aug 13)合成SA-T2序列。其中,SA为剪切受体,用于RNA的成熟剪切,应用转座子外显子捕获原理,在该位置与上游的第一的外显子完成拼接;T2A为翻译成氨基酸后会在细胞质中发生剪切形成释放抗性蛋白。其核苷酸序列如下:
SA-T2A(小写核酸序列为SA序列,大写核酸序列为T2A序列):
5’-ctgacctcttctcttcctcccacagggcctcgagagatctggcagcggaGAGGGCAGAGGAAGTCTGCTAACATGCGG TGACGTCGAGGAGAATCCTGGCCCA-3’。
1.2扩增PuroR和NeoR基因序列并与SA-T2A序列拼接:
以pSMPUW-IRES-Puro(Cell Biolabs)为模板,使用高保真酶(Phanta HS Super-Fidelity DNA Polymerase,诺唯赞生物公司)扩增PuroR基因,其中,所用引物PuroR-F和PuroR-R的核苷酸序列如下:
PuroR-F:5’-GGAGAATCCTGGCCCAATGACCGAGTACAAGCCCAC-3’;(SEQ ID NO.8)
PuroR-R:5’-GTTGGGCGCGCCTCATCCTGCAGTCAGGCACCGGGCTTGCGG-3’;(SEQ ID NO.9)
以pcDNA3.1(+)(Invitrogen)为模板,使用高保真酶(Phanta HS Super-Fidelity DNA Polymerase,诺唯赞生物公司)扩增NeoR基因,其中,所用引物NeoR-F和NeoR-R的核苷酸序列如下:
NeoR-F:5’-AGAATCCTGGCCCAATGATTGAACAAGATGGATTGCACG-3’;(SEQ ID NO.10)
NeoR-R:5’-AGGTTGGGCGCGCCGGCGTCGCTTGGTCGGTC-3’;(SEQ ID NO.11)
将扩增后的PuroR基因和NeoR基因分别与SA-T2A序列通过重叠PCR(overlap PCR)拼接,获得SA-T2A-PuroR和SA-T2A-NeoR。其中所用引物序列同上。其反应体系包括:100ng PuroR或NeoR分别作为模板,1μL(10μM)上游引物SA-T2A与1μL(10μM)PuroR-R或NeoR-R分别作为扩增引物,1μL DNA Polymerase,10μL 5x SF Buffer,1μL(10μM)dNTP Mix和32μL ddH2O。其反应条件为:95℃2min;95℃10s,54℃30s,72℃30s,10x循环;95℃10s,65℃20s,72℃20s,25x循环;72℃5min;4℃维持。
1.3扩增BGH PolyA信号序列,并分别与SA-T2A-PuroR和SA-T2A-NeoR拼接:
以pcDNA3.1(+)为模板,使用高保真酶(Phanta HS Super-Fidelity DNA Polymerase,诺唯赞生物公司)扩增BGH PolyA信号序列。其反应体系包括:1ng pcDNA3.1(+)作为模板,1μL(10μM)上游引物BGH-F与1μL(10μM)BGH-R分别作为扩增引物,1μL DNA Polymerase,10μL 5x SF Buffer,1μL(10μM)dNTP Mix和32μL ddH2O。其反应条件为:95℃10s,65℃20s,72℃20s,30x循环;72℃5min;4℃维持。其中,所用引物BGH-F和BGH-R的核苷酸序列如下
BGH-F:5’-GGCGCGCCCAACCTACGACTGTGCCTTCTAGTTGCCAGC-3’;(SEQ ID NO.12)
BGH-R:5’-CTAGCCATAGAGCCCACCGCATCCC-3’。(SEQ ID NO.13)
随后将SA-T2A-PuroR和SA-T2A-NeoR与BGH PolyA信号序列通过重叠PCR获得SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA(图4),其中包含SA,T2A,PuroR或NeoR和BGH PolyA信号。核苷酸序列分别如SEQ ID NO.1和SEQ ID NO.2所示。其反应体系中,模板为10ng SA-T2A-PuroR或SA-T2A-NeoR和10ng BGH PolyA,引物为1μL(10μM)上游引物SA-T2A与1μL(10μM)BGH-R,其余部分的体系与反应条件同步骤1.2中所述。
1.4使用T4多聚核苷酸激酶(购于NEB公司)处理SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA片段,使DNA片段5’末端磷酸化以用于后续的连接反应。
2.构建靶向AAVS1基因的Cas9/gRNA-AAVS1表达载体:
2.1使用BbsI内切酶(购于NEB公司)酶切CRISPR/Cas9表达载体(尧舜禹生物公司),并切胶回收DNA片段;
2.2参考文献(Hockemeyer et al Nat Biotechnol.2009Aug 13)所提供的靶向序列(GGGGCCACTAGGGACAGGAT),自行构建gRNA载体,设计并合成构建AAVS1-gRNA的两条寡核苷酸序列AAVS1-F和AAVS1-R,使用TE buffer溶解,并使用退火Buffer退火两段寡核苷酸序列。其反应体系包括:48μL退火缓冲液,1μL(100μM)AAVS1-F和1μL(100μM)AAVS1-R。使用PCR加热95℃5min,然后自然降温。其中,引物序列的核苷酸序列如下:
AAVS1-F:5’-CACCGGGGCCACTAGGGACAGGAT-3’;(SEQ ID NO.14)
AAVS1-R:5’-AAACATCCTGTCCCTAGTGGCCCC-3’。(SEQ ID NO.15)
2.3将退火产物与CRISPR/Cas9表达载体线性化片段使用T4连接酶(购于NEB公司)连接,并转化入DH5α细菌感受态,构建Cas9/gRNA-AAVS1表达载体。
3.构建靶向AAVS1同源重组载体:
以pGEM-T easy载体(购于Promega公司)为骨架载体,分别构建同源重组载体AAVS1-PuroR-BGH PolyA和AAVS1-NeoR-BGH PolyA(图5)。载体均含有剪切受体(splice adopter,SA)主要用于RNA的成熟剪切;T2A翻译成氨基酸后会在细胞质中发生剪切形成释放抗性蛋白;嘌呤霉素抗性基因(PuroR)或新霉素抗性基因(NeoR)应用于同源重组后的筛选;牛生长因子多聚腺苷酸信号(BGH PolyA)用于终止RNA的转录,AAVS1为左右同源重组臂,上下游各800bp。步骤如下:
3.1使用HEK293细胞(Homo sapiens embryonic kidney 293,ATCC(CRL-1573))基因组DNA为模板,使用所述引物AAVS1-LHA-F和AAVS1-RHA-R扩增AAVS1同源臂序列,使用PCR产物纯化试剂盒纯化后,并用Taq酶(购于诺唯赞生物公司)在DNA尾部加A,然后连接于pGEM-T easy载体(购于Promega公司),获得AAVS1-T载体。所用引物的核苷酸序列如下:
AAVS1-LHA-F:5’-CTGCTTTCTCTGACCTGCATTCTC-3’;(SEQ ID NO.16)
AAVS1-RHA-R:5’-AAGAGCAGAGCCAGGAACCCC-3’。(SEQ ID NO.17)
3.2以AAVS1-T载体为模板,以所述引物AAVS1-LHA-R和AAVS1-RHA-F扩增整个AAVS1-T载体,应用反向PCR将该载体在特定位置线性化(示意图见图5),并使用DpnI内切酶(购于NEB公司)处理PCR产物,去除质粒模板。所用引物的核苷酸序列如下:
AAVS1-LHA-R:5’-GCCCCACTGTGGGGTGGAG-3’;(SEQ ID NO.18)
AAVS1-RHA-F:5’-ACTAGGGACAGGATTGGTGAC-3’。(SEQ ID NO.19)
3.3将步骤1.4中所获得的磷酸化SA-T2A-PuroR-BGH PolyA和SA-T2A-NeoR-BGH PolyA分别与线性化的AAVS1-T载体连接,并转化入DH5α细菌感受态,构建AAVS1-PuroR-BGH PolyA和AAVS1-NeoR-BGH PolyA同源重组载体并测序。其AAVS1同源重组载体结构示意图见图5所示。
4.同源重组载体与Cas9/gRNA转染细胞与筛选。
4.1使用lip2000转染试剂(购于Gbico),按照试剂说明书,共转染步骤3.3中获得的线性化同源重组载体和步骤2.3中所获得的Cas9/gRNA-AAVS1表达载体。细胞培养使用MEM培养基(MEM培养基(购于Gbico)含10%FBS血清(购于Gbico)和1%青链霉素(购于Hyclone)),;使用胰蛋白酶(购于Gbico)消化细胞在6mm细胞培养皿接种2*106个细胞/每孔,24h后转染线性化同源重组载体共4μg(AAVS1-PuroR-BGH PolyA和AAVS1-NeoR-BGH PolyA载体)和4μg Cas9/AAVS1-gRNA载体。该含有双筛选标签载体分别与两条等位基因同源重组示意图见图6。
4.2转染24小时后,将细胞1:3进行传代。
4.3转染72小时后,加入1μg/mL浓度的嘌呤霉素(Sigma)进行筛选。
4.4加入嘌呤霉素筛选72小时后,加入400μg/mL的G418(Sigma)进行筛选2周,获取多克隆细胞株。
实施例2
本实例2提供了对本发明中转录终止信号SV40PolyA(Simian vacuolating virus 40PolyA)、BGH PolyA(bovine growth hormone PolyA)和β-globin teminator在其终止效率、分子量大小、以及反向序列的终止效率方面的优化比较(见图1)。其中,SV40PolyA(Simian vacuolating virus 40PolyA)、BGH PolyA(bovine growth hormone PolyA)是分子可隆重广泛应用的转录终止信号;而β-globin teminator是基因β-globin的终止信号,为研究较为透彻的转录终止信号。使用这三种转录终止信号,比较其终止效率,以及分子量大小,以及反向序列的终止效率见图1。
具体包括以下步骤:
a)针对SV40PolyA、β-globin teminator和BGH PolyA设计PCR扩增引物。SV40PolyA扩增以质粒----为模板;β-globin teminator以人类基因组为模板;BGH PolyA以pcDNA3.1为模板。获得的DNA片段通过EcoRI酶切位点插入pIRES-EGFP多克隆位点。分别构建BGH PolyA(+)、BGH PolyA(-)、SV40PolyA和β-globin见图1。所用BGH PolyA,SV40PolyA和β-globin引物序列如下:
BGH-PolyA-F:5’-TTCGAATTCCTGTGCCTTCTAGTTGCC-3’
BGH-PolyA-R:5’-CAGAATTCCCATAGAGCCCACCGCATC-3’
SV40PolyA-F:5’-GCTGAATTCTTAACTTGTTTATTGCAGCTTATAATGGTTAC-3’
SV40PolyA-R:5’-GCTGAATTCTAAGATACATTGATGAGTTTGGACAAACCA-3’
β-globin-F:5’-TTCGAATTCGCTCGCTTTCTTGCTG-3’
β-globin-R:5’-GCAGAATTCCTCCCACCCCCAAC-3’
b)将构建的载体分别转染HEK293细胞,24小时按照TRIzol RNA提取试剂(购于Life公司)说明书提取RNA,并按照PrimeScriptTM RT reagent Kit with gDNA Eraser(购于Takara)说明书逆转录提取的RNA,并使用RT-qPCR检测IRES的转录表达水平,NeoR转录为内参见图2。此检测方法中所使用的引物的核苷酸序列如下:
IRES-F:5’-CCCTGTCTTCTTGACGAGCAT-3’
IRES-R:5’-GGGTCGCTACAGACGTTGTTT-3’
NEO-F:5’-ATCCATCATGGCTGATGCAATGCG-3’
NEO-R:5’-CCATGATATTCGGCAAGCAGGCAT-3’
结果(图3)显示三个终止信号的BGH PolyA均有良好的终止效率,其中BGH PolyA和β-globin终止效率最高,同时BGH PolyA反向仅有微弱的终止效率见。三个终止信号的大小分别为:BGH-PolyA:255bp,SV40PolyA:122bp,β-globin teminator:1753bp。考虑到转录终止的效率与分子量的大小,优选BGH PolyA作为本发明的终止信号。
实施例3
本实施例3中,使用有限稀释法从实施例1中所获得的多克隆细胞株中获得单克隆细胞株,并通过RT-qPCR检测PPP1R12C基因的转录情况。
具体包括如下步骤:
a)在使用有限稀释法获得单克隆细胞株时,先对实施例1中筛选后获得的细胞进行稀释。10ml培养基中约50个细胞,96孔板中每个孔接种100μl,培养15天;
b)分别按照TRIzol试剂(购于Life公司)说明书提取DNA和RNA;
c)采用基因分型PCR方法,以同源重组区域的上游和下游引物如图7所示,以提取样本DNA为模板,进行PCR检测(图8)。此检测方法中所使用的引物的核苷酸序列如下:
AAVS1-HDR-F:5’-ATTGTCACTTTGCGCTGCCC-3’
AAVS1-HDR-R:5’-CCTCTCGTGGGGTCCAGG-3’。
其中含有多克隆基因组与未转染基因组的DNA作为模板的样本1中,扩增出三条带:3654bp为Neo重组后等位基因扩增条带,3310bp为PuroR重组后等位基因扩增条带,1976bp为野生型等位基因扩增条带;然而,在以使用本发明的方法终止基因转录后的单克隆细胞系基因组DNA为模板的样本2中,其扩增结果仅有3654bp和3310bp,无野生型条带。比较图8中的样本1与样本2可以看出,在单克隆中两条等位基因均插入了含有不同抗生素基因的PolyA序列,且不存在含有野生型(即1976bp序列)的等位基因。
d)按照PrimeScriptTM RT reagent Kit with gDNA Eraser(购于Takara)说明书逆转录提取的RNA,并使用SYBR Premix Ex Taq(购于Takara)进行RT-qPCR检测PPP1R12C RNA的表达水平,GAPDH为内参。此检测方法中所使用的引物的核苷酸序列如下(Primer Express3.0,购于ABI公司):
PPP1R12C-F:5’-ATCGCCGCCGTCAACAGT-3’;
PPP1R12C-R:5’-CCCATTCAGCCAGCACCTC-3’;
GAPDH-F:5’-GACTCCACGACGTACTCAGCGCCAGCATCG-3’;
GAPDH-R:5’-AAGGTGAAGGTCGGAAGGTGAAGGTCGG-3’。
按照c)步骤中的方法挑选的双等位基基因均插入的PolyA的阳性克隆株。挑选三个克隆,clone1-3,检测其PPP1R12C RNA的表达水平检测结果如图9所示。相对于未插入PolyA的对照细胞,BGH PolyA的三个克隆显著的降低了PPP1R12C信使RNA的表达水平。每个样本下降水平均在99%以上。该结果显示,等位基因均插入BGH PolyA,能够有效的终止PPP1R12C基因的转录,极为显著的降低信使RNA的表达水平。
实施例4
本实施例4中,使用有限稀释法从实施例1中所获得的多克隆细胞株中获得单克隆细胞株,通过基因分型PCR评估了双等位基因插入BGH PolyA的插入效率。
具体包括如下步骤:
a)采用上述实施例3中a)中方法获取大量单克隆细胞;
b)单克隆细胞按照TRIzol试剂(购于Life公司)说明书提取DNA;
c)采用上述实施例3中c)中方法,以单克隆DNA为模板评估基因组插入效率。
表1显示了针对单克隆样本提取基因组DNA进行双等位基因插入的基因型分析结果:在125单克隆样本中,发生双等位基基因插入的单克隆为118个,而单个等位基因插入的单克隆为7个,均为嘌呤霉素插入序列。使用含有双抗性筛选基因的同源重组载体同时整合入等位基因中,经过双抗生素的筛选能够高效的获得双等位基因插入的阳性克隆,其效率高达94.4%。结果显示使用含有两种抗生素基因的PolyA的方法,能够极为高效的筛选双等位基因均插入的细胞株。而插入片段的PolyA序列则高效的终止基因的转录,极为显著的降低信使RNA的表达水平。
表1.双等位基因BGH PolyA的插入效率
注:细胞等位基因通过同源重组分别插入含有不同抗性基因的BGH PolyA序列,检测获得单克隆细胞株的同源重组效率。
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,本发明要求保护范围由所附的权利要求书、说明书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!