吲哚酮螺四氢硫代吡喃类衍生物及其制备方法和应用与流程
本发明涉及医药技术领域,具体涉及一类新型吲哚酮螺四氢硫代吡喃类衍生物及其制备方法,以及在抗肿瘤方面的应用。
背景技术:
螺吲哚酮骨架作为天然产物和活性分子的优势骨架,受到了有机合成和药物化学研究人员的广泛关注。目前,已经报道了多样性的螺吲哚酮类化合物,并发现具有广泛的药理活性,如孕激素受体调节剂(Jay E.Wrobel et al,Design,Synthesis,and SAR of New Pyrrole-Oxindole Progesterone Receptor Modulators Leading to 5-(7-Fluoro-3,3-dimethyl-2-oxo-2,3-dihydro-1H-indol-5-yl)-1-methyl-1H-pyrrole-2-carbonitrile(WAY-255348),J.Med.Chem.2008,51:1861-1873)、抗艾滋病(Singh RK.et al,Rhodium(II)acetate-catalyzed stereoselective synthesis,SAR and anti-HIV activity of novel oxindoles bearing cyclopropane ring,Eur.J.Med.Chem.2011,46:1181-1188)、抗肿瘤(WO2008055812A1)、抗结核病(Waldmann,H.et al,Identification of thiazolidinones spiro-fused to indolin-2-ones as potent and selective inhibitors of the Mycobacterium tuberculosis protein tyrosine phosphatase B,Angew.Chem.Int.Ed.Engl.2010,49:5902-5905)、抗疟疾(Diagana,T.T.et al,Spiroindolones,a potent compound class for the treatment of malaria,Science 2010,329:1175-1180)和MDM2抑制剂(Wang,S.et al,Structure-based design of potent non-peptide MDM2inhibitors,J.Am.Chem.Soc.2005,127:10130-10131)等。
本发明人的前期研究,构建了一类吲哚酮螺四氢硫代吡喃类衍生物,并发现其具有很好的体外抗肿瘤活性(申请号:CN201410233972.5;公布号CN103992334A,DOI:10.1021/acs.orglett.6b00155)。在此研究基础上,我们进一步构建了一类新型吲哚酮螺四氢硫代吡喃类衍生物,较前期研究成果相比,这些衍生物具有全新的骨架结构,并可以通过控制有机催化剂的构型得到d-型或l-型的异构体;而且这类衍生物具有较强的抗肿瘤活性,可以进行抗肿瘤药物的开发。
技术实现要素:
本发明的目的是提供一类具有全新骨架结构的吲哚酮螺四氢硫代吡喃类衍生物,本发明的另一目的是提供该类吲哚酮螺四氢硫代吡喃类衍生物的制备方法,本发明的第三目的是提供该类吲哚酮螺四氢硫代吡喃类衍生物的应用。
本发明的第一方面,是提供一类吲哚酮螺四氢硫代吡喃类衍生物,包括其消旋体,以及d-型或l-型异构体,所述的化合物的结构如通式I所示:
其中,
R1表示:苯环上的各种取代基;其中苯环上的取代基可以是单取代,也可以是多取代,取代基团表示:氢、卤素、低级卤代烷基、低级烷基、羟基、低级羟基烷基、低级烷氧基、氨基、低级烷基氨基、低级卤代烷基氨基、低级环烷基氨基、低级链炔基氨基、硝基、低级硝基烷基、氰基、低级氰基烷基、酰胺基、低级环烷基酰胺基、低级酰胺基烷基。优选R1表示下列基团:氢、卤素、低级烷基、羟基、低级烷氧基、氨基、低级烷基氨基、低级环烷基氨基、硝基、酰胺基、低级环烷基酰胺基。
R2表示:各种芳环取代基团。芳环可以为苯环或取代苯环,也可以为各种杂环如呋喃、噻吩、吡啶等。芳环上可以含有各种取代基团,如氢、卤素、低级烷基、羟基、低级烷氧基、氨基、低级烷基氨基、低级环烷基氨基、硝基、酰胺基、低级环烷基酰胺基等;优选R2表示下列基团:苯环、卤素单取代或双取代的苯环、烷氧基取代的苯环。
R3表示:各种芳环,芳环可以为苯环或取代苯环,也可以为各种杂环,如吡咯、呋喃、噻吩、吡啶等;芳环上可以含的取代基团为氢、卤素、低级烷基、羟基、低级烷氧基、氨基、低级烷基氨基、低级环烷基氨基、硝基、酰胺基、低级环烷基酰胺基;优选R3表示下列基团:苯环、卤素单取代或双取代的苯环、烷基或烷氧基取代的苯环、呋喃。
R4表示:硝基、氨基或者酰胺基团;优选NO2或NH2。
R5表示:氢或低级烷基或低级羰基侧链;优选H或Ac。
所述的“低级环烷基”是指含3至7个碳的环,其它所述的“低级”取代基是指相应的脂肪烃基是直链或支链的、饱和的、并且含1至6个碳原子。
上述吲哚酮螺四氢硫代吡喃类衍生物可以为其消旋体,也可以为其d-型或l-型异构体。
经试验抗肿瘤效果较好的本发明的优选化合物各基团的组合详见表1,核磁、质谱等数据见表2。
表1本发明部分优选化合物的化学结构
发明的第二方面,是提供了上述的一类吲哚酮螺四氢硫代吡喃类衍生物,包括其消旋体、d-型或l-型异构体的制备方法。
本发明的化合物的反应流程如下:
目标化合物的制备:
a)中间体II的制备参照文献报道的方法(Advanced Synthesis&Catalysis2013,355:829-835)。将硝基甲烷(7.2mL,0.134mol)、取代的苯甲醛(原料I,0.022mol)和醋酸铵(0.20g,2.6mmol)加入到80mL的乙酸中,并在100℃下反应10h。反应完全后,蒸干溶剂,残留物柱层析纯化(石油醚:乙酸乙酯=100:3),得到中间体II。
b)中间体III的制备参照文献报道的方法(Advanced Synthesis&Catalysis2013,355:829-835)。在50mL的二氯甲烷中,加入中间体II(14.0mmol)、乙酰氯(2.1mL,29.6mmol)和无水三氯化铁(4.64g,28.6mmol),室温下反应3h。反应完全后,蒸干溶剂,残留物柱层析纯化(DCM:MeOH=100:1),得到中间体III。
c)中间体V的制备
在氮气保护下,将中间体III、中间体IV、三乙胺加入到二氯甲烷中,并室温搅拌反应,反应完全后,蒸干溶剂,柱层析纯化,DCM:MeOH=100:1,得到中间体V。
d)目标化合物VIII的制备
在二氯甲烷中,依次加入催化剂VII、中间体V、中间体VI,室温下搅拌12h,反应完全后,蒸干溶剂,残留物经柱层析纯化,洗脱剂为DCM,得到目标产物VIII;
所述的催化剂VII为一种氨基硫脲类双功能催化剂,CAS号为1448608-06-7,但催化剂并不局限于这一种,也可以为其他的双功能催化剂(分子结构中同时含有氨基(伯胺、仲胺或叔胺)、硫脲或脲基团,代表性催化剂如下式所示,本发明专利以VII为例说明)。
e)目标化合物IX的制备
在异丙醇中,加入化合物VIII、锌粉、盐酸和醋酸,在室温下反应完全后,柱层析纯化,洗脱剂为DCM:MeOH=100:3,得到目标产物IX。
f)目标化合物X的制备
在二氯甲烷中,加入化合物VIII、溴代烷烃或酸酐和适量催化剂,在常温或加热条件下反应完全后,柱层析纯化,洗脱剂为PE:EtOAc=5:1,得到目标产物X。
本发明的化合物可通过采用不同构型的催化剂VII制备目标化合物VIII的不同异构体。
以化合物1为例说明(如以下反应流程式):采用消旋体催化剂VII,制备得到消旋体的目标化合物1。采用光学纯VII(CAS:1448608-06-7)为催化剂,则得到(2'R,3'R,4'R,5'R)-型目标化合物VIII。采用VII的对映异构体(CAS:1448608-07-8)为催化剂,则得到(2'S,3'S,4'S,5'S)-型目标化合物VIII。
本发明的第三方面,是提供了还提供了上述的一类吲哚酮螺四氢硫代吡喃类衍生物,包括其消旋体、d-型或l-型异构体,在制备抗肿瘤药物中的应用。
所述的肿瘤为肺癌、肠癌、乳腺癌、肝癌等。
对本发明的化合物进行了肿瘤细胞增殖抑制试验,试验方法采用常规的MTT法,细胞株选用A549(人肺癌细胞)和MCF-7(人乳腺癌细胞)。培养液为DMEM+15%NBS+双抗。以上实验结果表明,本发明的化合物具有良好的抗肿瘤活性,部分化合物的抗肿瘤活性与对照药Nutlin-3(CAS:548472-68-0)相当,因此本发明化合物可以用于制备抗肿瘤药物。
本发明合成的化合物具有全新的骨架结构,本发明的化合物含有硝基、羟基、氨基等基团,有利于快速衍生化;此外本发明通过控制手性催化剂的构型,可以得到不同构型的目标产物。本发明提供了新型的抗肿瘤候选药物。
附图说明
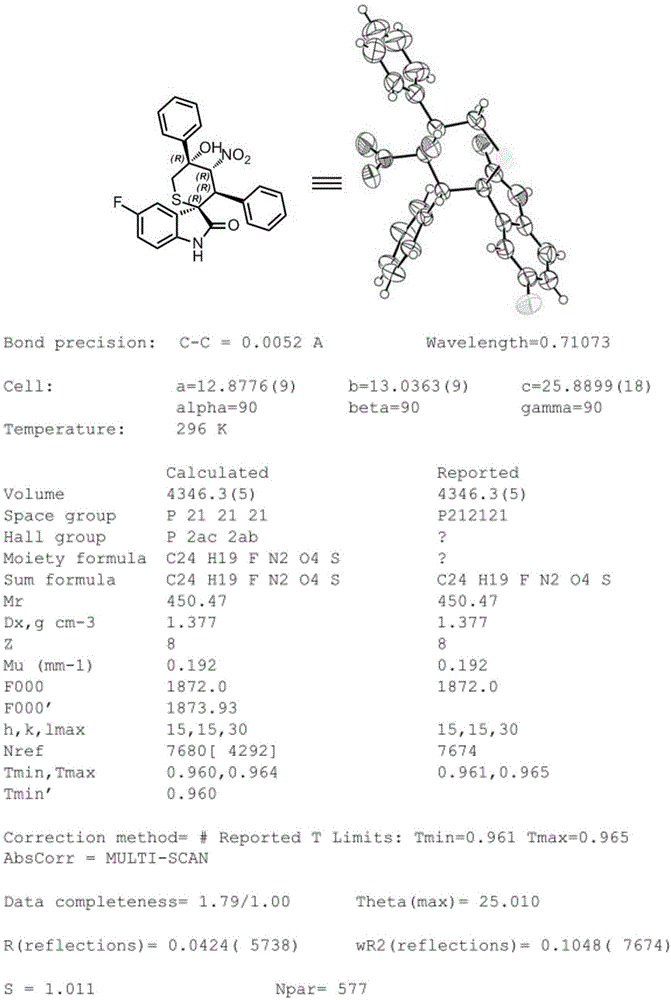
图1为化合物14的X单晶衍射数据。
图2为化合物1的ee值测定,上图为化合物1的消旋体液相图谱,下图为光学纯化合物1的液相图谱。液相条件与结果为:HPLC(Chiralpak OD,0.46cm I.D.×25cm L×5um,25℃,异丙醇/正己烷=15:85,流速0.8mL/min,λ=254nm):tmajor=12.73min,tminor=15.52min,ee=94%。
具体实施方式
现结合实施例和附图,对本发明作详细描述,但本发明的实施不仅限于此。本发明所用试剂和原料均市售可得或可按文献方法制备。
下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
以下实施例所涉化合物对应通式Ⅰ的化学结构式、1H-NMR、13C-NMR和HRMS数据详见表2,其中编号1-18分别对应表1中的化合物1-18、表3中的化合物1-18、实施例1-18。
表2本发明部分优选化合物的1H-NMR,13C-NMR和HRMS数据
实施例1:化合物1的合成
A中间体(E)-1-溴-2-(2-硝基乙烯基)苯的制备(中间体II)
参照文献报道的方法(Advanced Synthesis&Catalysis 2013,355:829-835)。将硝基甲烷(7.2mL,0.134mol)、2-溴苯甲醛(4.0g,0.022mol)和醋酸铵(0.20g,2.6mmol)加入到80mL的乙酸中,并在100℃下反应10h。反应完全后,蒸干溶剂,残留物柱层析纯化(石油醚:乙酸乙酯=100:3),得到(E)-1-溴-2-(2-硝基乙烯基)苯为黄色固体4.21g,收率为85.4%。
B中间体4-溴-3-氯吲哚-2-酮的制备(中间体III)
参照文献报道的方法(Advanced Synthesis&Catalysis 2013,355:829-835)。在50mL的二氯甲烷中,加入(E)-1-溴-2-(2-硝基乙烯基)苯(3.2g,14.0mmol)、乙酰氯(2.1mL,29.6mmol)和无水三氯化铁(4.64g,28.6mmol),室温下反应3h。反应完全后,蒸干溶剂,残留物柱层析纯化(DCM:MeOH=100:1),得到4-溴-3-氯吲哚-2-酮为黄白色固体2.25g,收率为65.6%。
C中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮的制备(中间体V)
在20mL的二氯甲烷中,加入4-溴-3-氯吲哚-2-酮(0.3g,1.22mmol)、2-巯基-1-苯乙酮(0.24g,1.59mmol)和0.1mL的三乙胺,氮气保护下室温反应过夜。反应完全后,蒸干溶剂,残留物柱层析纯化(DCM:MeOH=100:1),得到中间体V为黄白色固体0.27g,收率为62%。1H NMR(400MHz,CDCl3)δ4.11(d,J=15.5Hz,1H),4.34(d,J=15.5Hz,1H),4.48(s,1H),6.82(d,J=7.4Hz,1H),7.10-7.17(m,2H),7.46(t,J=7.7Hz,2H),7.59(t,J=7.4Hz,1H),7.93(d,J=8.0Hz,2H),8.37(s,1H).13C NMR(100MHz,CDCl3)δ36.13,45.49,108.95,120.63,124.97,126.57,128.60(2C),128.68(2C),130.84,133.52,135.55,142.76,175.76,194.37.HRMS(ESI+)m/z calculated for C16H13BrNO2S(M+H):361.9850,found 361.9848.
D目标化合物1的制备
在2mL的DCM中,依次加入催化剂VII(CAS:1448608-06-7,0.01mmol)、中间体V(0.10mmol)和VI(0.12mmol),室温下搅拌反应12h。反应完全后,蒸干溶剂。残留物经柱层析纯化(DCM)得到目标化合物1--(2'R,3'R,4'R,5'R)-型目标化合物VIII,为白色固体(39mg),收率为76%,ee值为94%(化合物1的ee值测定如图2所示),其中四个手性中心的构型都为R型。
化合物2-16的合成参照化合物1的合成方法。
实施例2:化合物2的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与4-溴-β-硝基苯乙烯(0.12mmol)反应,制备化合物2为白色固体(42mg),收率74%,ee值为91%。
实施例3:化合物3的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与3-氟-β-硝基苯乙烯(0.12mmol)反应,制备化合物3为白色固体(35mg),收率66%,ee值为89%。
实施例4:化合物4的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与2-氟-β-硝基苯乙烯(0.12mmol)反应,得到化合物4为白色固体(32mg),收率61%,ee值为96%。
实施例5:化合物5的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与3-溴-β-硝基苯乙烯(0.12mmol)反应,得到化合物5为白色固体(42mg),收率72%,ee值为95%。
实施例6:化合物6的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与4-甲基-β-硝基苯乙烯(0.12mmol)反应,制备化合物6为白色固体(34mg),收率65%,ee值为95%。
实施例7:化合物7的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与4-甲氧基-β-硝基苯乙烯(0.12mmol)反应,制备化合物7为白色固体(42mg),收率77%,ee值为92%。
实施例8:化合物8的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与(E)-2-(2-硝基乙烯基)呋喃(0.12mmol)反应,制备化合物8为白色固体(29mg),收率58%,ee值为87%。
实施例9:化合物9的合成
参照实施例1。中间体4-溴-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与3,5-二氟-β-硝基苯乙烯(0.12mmol)反应,制备化合物9为白色固体(39mg),收率72%,ee值为87%。
实施例10:化合物10的合成
参照实施例1。中间体4-溴-3-((2-(4-甲氧基苯基)-2-氧代乙基)硫)吲哚-2-酮(0.10mmol)与3-氟-4-溴-β-硝基苯乙烯(0.12mmol)反应,制备化合物10为白色固体(49mg),收率77%,ee值89%。
实施例11:化合物11的合成
参照实施例1。中间体4-溴-3-((2-(4-甲氧基苯基)-2-氧代乙基)硫)吲哚-2-酮(0.10mmol)与β-硝基苯乙烯(0.12mmol)反应,得到化合物11为白色固体(30 mg),收率56%,ee值90%。
实施例12:化合物12的合成
参照实施例1。中间体4-溴-3-((2-(4-溴苯基)-2-氧代乙基)硫)吲哚-2-酮(0.10mmol)与β-硝基苯乙烯(0.12mmol)反应,得到化合物12为白色固体(38mg),收率65%,ee值92%。
实施例13:化合物13的合成
参照实施例1。中间体4-氯-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与β-硝基苯乙烯(0.12mmol)反应,制备化合物13为白色固体(34mg),收率73%,ee值94%。
实施例14:化合物14的合成
参照实施例1。中间体5-氟-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与β-硝基苯乙烯(0.12mmol)反应,得到化合物14为白色固体(32mg),收率71%,ee值94%。
实施例15:化合物15的合成
参照实例1。中间体3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与β-硝基苯乙烯(0.12mmol)反应,得到化合物15为白色固体(28mg),收率65%,ee值97%。
实施例16:化合物16的合成
参照实施例1。中间体6-甲基-3-((2-氧代-2-苯乙基)硫)吲哚-2-酮(0.10mmol)与β-硝基苯乙烯(0.12mmol)反应,得到化合物16为白色固体(25mg),收率56%,ee值93%。
实施例17:化合物17的合成
在2mL异丙醇中,加入化合物1(20mg)、Zn粉25mg、乙酸0.2mL和10%的盐酸0.3mL,并在常温下反应完8h。反应完全后,柱层析纯化,洗脱剂为DCM:MeOH=100:3得到化合物17为白色固体13mg,收率为66%,ee值为94.7%。
实施例18:化合物18的合成
在3mL二氯甲烷中加入化合物1(20mg)、4-二甲胺基吡啶3mg、乙酸酐4.4mg,室温下反应1h,反应完全后蒸干溶剂,柱层析纯化,洗脱剂为PE:EtOAc=5:1,得到化合物18为白色固体(19mg),收率为88%.
实施例19:化合物1的消旋体制备
在2mL的二氯甲烷中,依次加入消旋体催化剂VII(CAS:1448608-06-7与CAS:1448608-07-8两种对应异构体等量混合)、中间体V和VI,室温下搅拌反应12h。反应完全后,蒸干溶剂。残留物经柱层析纯化(DCM)得到消旋体目标化合物1为白色固体(39mg),收率为76%。
其它消旋体目标化合物的制备参照实施例19。
实施例20:化合物1的对映异构体制备
在2mL的DCM中,依次加入催化剂VII的对映异构体(CAS:1448608-07-8)、中间体V和VI,室温下搅拌反应12h。反应完全后,蒸干溶剂。残留物经柱层析纯化(DCM)得到目标化合物1--(2'S,3'S,4'S,5'S)-型目标化合物VIII,为白色固体(39mg),收率为76%,四个手性中心的立体构型全部为S构型。
其它对映异构体的制备参照实施例20。
实施例21:本发明化合物的抗肿瘤活性试验
对本发明的化合物进行了肿瘤细胞增殖抑制试验,试验方法采用常规的MTT法(如吕秋军主编《新药药理学研究方法》,2007:242-243)。
细胞株选用肺癌A549、乳腺癌MCF-7,均购自上海市医药工业研究院。
培养液为DMEM+15%NBS+双抗。
样品液配制:用DMSO(Merck)溶解后,加入PBS(-)配成100μg/mL的溶液或均匀的混悬液,然后用DMSO的PBS(-)稀释,最终浓度分别为10μg/mL、1μg/mL、0.1μg/mL、0.01μg/mL、0.001μg/mL、0.0001μg/mL。
将抗肿瘤化合物Nutlin-3以同样的条件配成对照品溶液。
96孔板每孔加入浓度为3×104个/mL的细胞悬液100μL,即3000个细胞/孔,置37℃、5%CO2培养箱内。24小时后,分别加入样品液和对照品液,10μL/孔,37℃作用72小时。每孔加入5mg/mL的MTT(3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑翁溴化物)溶液20μL,作用4小时后加入溶解液DMSO,100μL/孔,置培养箱内,次日用MK-2全自动酶标仪测570nm OD值,计算半数抑制浓度IC50。
部分优选化合物的抗肿瘤活性详见表3,其中,样品1-18是指相应实施例中制备的吲哚酮螺四氢硫代吡喃类化合物,如化合物1表示在实施例1中所得到的化合物,同理类推。1a代表化合物1的消旋体,1b代表化合物1的对映异构体。其中化合物19为专利CN201410233972.5中活性最优的化合物,结构如表3所示。
结果显示,本专利申请的化合物总体表现出广谱、中等的抗肿瘤活性,部分化合物的活性与对照化合物19相当。其中化合物2与阳性药Nutlin-3(CAS:548472-68-0)表现相当的抗肿瘤活性,优于之前申请的化合物19,可以作为抗肿瘤的先导结构进行更加深入的研究。此外,本专利申请的化合物含有硝基、羟基、氨基等易于衍生化的基团,便于快速衍生化,发现活性优良的抗肿瘤先导化合物。
表3本发明部分化合物对肿瘤细胞的半数抑制浓度IC50(单位:μM)
a化合物1的消旋体;b化合物1的对映异构体;CPT为喜树碱
以上实验结果表明,本发明的化合物具有良好的抗肿瘤活性,因此本发明化合物可以用于制备抗肿瘤药物。
以上已对本发明创造的较佳实施例进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明创造精神的前提下还可作出种种的等同的变型或替换,这些等同的变型或替换均包含在本申请权利要求所限定的范围内。
- 还没有人留言评论。精彩留言会获得点赞!