一种壳寡糖或其衍生物修饰的聚氨酯纤维敷料及制备方法与流程
本发明属于生物医用材料技术领域,特别涉及一种壳寡糖或其衍生物修饰的聚氨酯纤维敷料及制备方法。
背景技术:
创面愈合是创伤外科中的一个重要课题,研究开发理想的促进创面愈合的材料备受关注。传统敷料如纱布、棉花等易滋生细菌,粘连伤口,更换时带来二次创伤。因此,国内外学者近年来大力开展新型敷料的研究,试图避免传统敷料的缺点。通常,创伤敷料不仅要求具有良好的生物相容性、吸液保液性、适宜的力学强度和弹性,还应具有隔菌作用,以防止创面的感染,而且能够促进创面的愈合。聚氨酯材料因其安全、无毒、无刺激、不致敏、良好的生物相容性和力学性能等特性,是目前国内外研究报道较多的一类创伤敷料。然而,单一的聚氨酯医用敷料不能有效地阻止创面被感染、加快创面修复,且容易与创面发生粘连,易造成二次创伤。近年,围绕改善聚氨酯敷料各种性能的相关专利和文献有不少报道。
申请号201510994613.6的中国发明专利公开了一种促进皮肤创面修复的敷料及制备方法,该敷料的组成及含量如下:聚氨酯20~30,九子连环草4~9,骨碎补3~6,连翘8~14,当归5~12,陆英4~8,积雪草6~10,地肤子8~12,兰香草9~13,赤芍6~9,黄芪4~7,聚乙二醇12~17,丙三醇9~11,磺胺7~10。该敷料具有促进创面局部血液循环,减轻创面局部肿胀等特性。又如申请号201510490782.6的中国发明专利公开了一种复合胶原的聚氨酯基双层敷料,该敷料具有可促进创面修复和减少疤痕形成等优点。
目前对聚氨酯敷料材料的改性主要集中在采用共混或复合等物理改性方法,引入的共混填料在基体中的分散性、稳定性等始终是存在的主要问题。点击化学因其反应条件温和、快速、有效和高产率等特点,近年成为材料科学家手中强有力的研究工具,并逐渐应用到各类聚合物材料的改性修饰等方面。若能灵活地利用点击化学这一先进技术对聚氨酯敷料在分子水平进行设计和生物学修饰,这必将为新型聚氨酯敷料的开发与应用提供一条有效而全新的途径。
壳寡糖是由壳聚糖经水解后产生的、由2~10个葡氨糖通过β-1-4糖苷键连接而成的低聚糖,其相对分子质量小、水溶性好、可被组织吸收利用。壳寡糖不仅具有壳聚糖优异的生物学功能,如良好的抗菌、止血、抗氧化、抗癌等生理活性,可以促进细胞分化和生长,促进创伤上皮化,形成肉芽组织,加速伤口的愈合,以及能够抑制纤维结缔组织,减少疤痕组织形成和促进血管内皮细胞生长等;而且,壳寡糖的生物学功能往往明显优于壳聚糖。因此,壳寡糖及其相关衍生物成为近年国内外研究和产品开发的热点。
技术实现要素:
为了克服上述现有技术的缺点与不足,本发明的首要目的在于提供一种壳寡糖或其衍生物修饰的聚氨酯纤维敷料的制备方法。本发明方法通过设计材料的制备路线、灵活运用点击化学,在温和的条件下高效获得一类壳寡糖或其衍生物修饰的聚氨酯纤维敷料。
本发明另一目的在于提供上述方法制备的壳寡糖或其衍生物修饰的聚氨酯纤维敷料。
本发明的目的通过下述方案实现:
一种壳寡糖或其衍生物修饰的聚氨酯纤维敷料的制备方法,包括以下步骤:
步骤一:采用聚乙二醇引发炔基化交酯单体开环聚合,合成侧链含炔基的生物可降解聚酯醚;
步骤二:以侧链含炔基的生物可降解聚酯醚为软段,与二异氰酸酯和扩链剂反应合成侧链含炔基的生物可降解聚氨酯;
步骤三:合成叠氮化壳寡糖或其衍生物;
步骤四:侧链含炔基的生物可降解聚氨酯与叠氮化壳寡糖或其衍生物发生点击化学反应,合成壳寡糖或其衍生物修饰的聚氨酯;
步骤五:通过静电纺丝技术构建壳寡糖或其衍生物修饰的聚氨酯纤维敷料。
步骤一中,所述合成侧链含炔基的生物可降解聚酯醚的聚合条件优选为:催化剂选用辛酸亚锡,催化剂用量为炔基化交酯单体用量的0.01~0.3mol%,反应温度为80~150℃,反应时间为6~54h。
所述聚乙二醇与炔基化交酯的摩尔投料比为:1:5~1:50。
所述聚乙二醇的数均分子量范围为:200~4000。
所述炔基化交酯单体的结构通式如(I)所示:
其中,R1、R2、R3和R4可相同或不同的分别为氢原子、甲基或炔基,且R1、R2、R3和R4至少有一个是炔基。
步骤二中,所述侧链含炔基的生物可降解聚氨酯的合成,是通过侧链含炔基的生物可降解聚酯醚与二异氰酸酯以及扩链剂反应得到,制备方法采用常规聚氨酯制备的一步法或两步法即可,具体条件可为:
所述侧链含炔基的生物可降解聚酯醚、二异氰酸酯以及扩链剂的摩尔投料比为:1:(2~10):(1~6);
所述的二异氰酸酯优选为六亚甲基二异氰酸酯、二环己基二异氰酸酯、甲苯二异氰酸酯等;
所述的扩链剂优选为乙二醇、1,4-丁二醇、一缩二乙二醇、乙二胺、1,4-丁二胺等。
步骤三中,所述的叠氮化壳寡糖或其衍生物是通过壳寡糖或其衍生物与叠氮化钠反应制备得到,其制备方法为本领域常规方法即可,具体可包括如下步骤:(1)将壳寡糖或其衍生物溶于水中,再加入十二烷基磺酸钠,两者摩尔比为1:1~1:10,在室温下反应0.5~8h,冷冻干燥得到烷基磺酸化的壳寡糖或其衍生物。(2)将烷基磺酸化壳寡糖或其衍生物溶于二甲基甲酰胺,加入对甲基苯磺酰氯,两者摩尔比为1:1~1:30,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖或其衍生物质量的0.5~5%,氮气保护下于-20-40℃反应4~12h,得到产物。(3)在二甲基甲酰胺中,按摩尔比1:1~1:50的投料比加入步骤(2)的反应产物和叠氮化钠,于60~120℃反应2~12h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖或其衍生物。
所述的壳寡糖或其衍生物优选为壳寡糖、壳寡糖季铵盐。
步骤四中,所述侧链含炔基的生物可降解聚氨酯与叠氮化壳寡糖或其衍生物发生点击化学反应的具体操作可包括如下步骤:将侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖或其衍生物溶于有机溶剂中,两者的摩尔比为20:1~1:20;将上述混合溶液加入到含催化剂的有机溶剂中,常温避光反应6~36h,得到所述壳寡糖或其衍生物修饰的聚氨酯。
所述的催化剂可为氯化亚铜(CuBr),或者是摩尔比为1:1的硫酸铜和抗坏血酸混合物中的一种。
所述催化剂的用量优选为侧链含炔基的生物可降解聚氨酯摩尔量的1~20%。
所述的有机溶剂可为四氢呋喃、N,N-二甲基甲酰胺和二甲基亚砜中的至少一种。
步骤五中,所述静电纺丝技术构建壳寡糖或其衍生物修饰的聚氨酯纤维敷料的制备方法如下:将壳寡糖或其衍生物修饰的聚氨酯溶解于有机溶剂中,得到质量体积浓度为5~20%的电纺丝溶液,在10~30kV的静电压下进行纺丝。
所述的有机溶剂可为四氢呋喃、N,N-二甲基甲酰胺和二甲基亚砜中的至少一种。
本发明提供上述方法制备得到的壳寡糖或其衍生物修饰的聚氨酯纤维敷料。本发明方法通过化学键的结合将具有多种生物功能性的壳寡糖或其衍生物引入到聚氨酯敷料中,从而赋予本发明敷料优异的亲水性、抗菌、消炎、止血、促进细胞分化与创面组织形成等生物功能性,其不仅具有良好的柔韧性和抗张力、适宜的多孔结构、良好的吸水和透气性,而且,具有控制细菌感染和促进创面修复的生物学功能,因此,本发明的壳寡糖或其衍生物修饰的聚氨酯纤维敷料可望得以广泛应用。
本发明相对于现有技术,具有如下的优点及有益效果:
(1)本发明采用简单、易行的技术路线设计合成壳寡糖或其衍生物修饰的生物可降解聚氨酯,并实现壳寡糖或其衍生物在生物降解聚氨酯侧链的分布以及密度可调控。
(2)由于点击化学反应的高度专一性和反应条件温和,本发明中无需对壳寡糖或其衍生物上的其它基团进行保护。
(3)本发明在温和的条件下,通过化学键的结合将具有多种生物功能性的壳寡糖或其衍生物引入到聚氨酯敷料中,从而赋予聚氨酯敷料优异的亲水性、抗菌、消炎、止血、促进细胞分化与创面组织形成等生物功能性。
(4)本发明涉及的制备条件温和,制备的新型聚氨酯纤维敷料不仅具有良好的柔韧性和抗张力、适宜的多孔结构、良好的吸水和透气性,而且,具有控制细菌感染和促进创面修复的生物学功能,因此,所获得的聚氨酯纤维敷料可望得以广泛应用。
附图说明
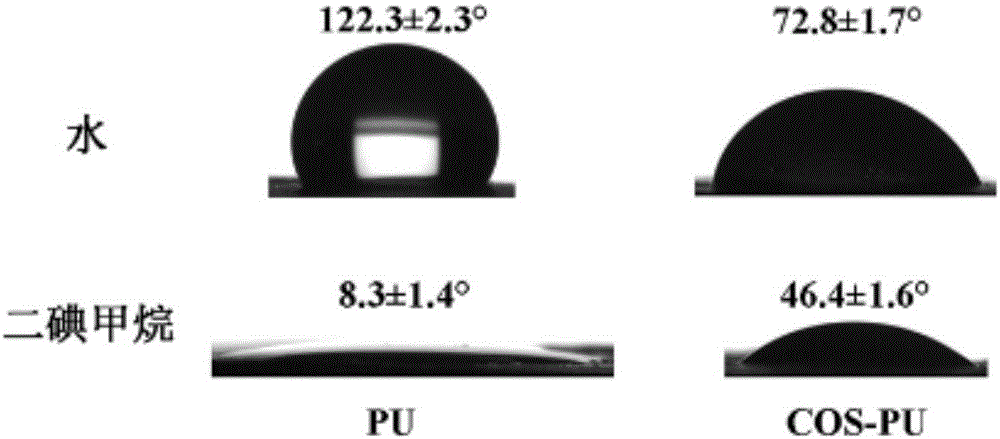
图1为本发明实施例1中所制备的PU和COS-PU纤维敷料的表面接触角。
图2为本发明实施例5中所制备的PU和COS-PU纤维敷料表面成纤维细胞细胞粘附的形貌与细胞核分布的激光共聚焦照片。
图3为本发明实施例6中所制备的PU和G-COS-PU纤维敷料表面成纤维细胞粘附的形貌的扫描电镜照片。
图4为本发明实施例8中所制备的PU和G-COS-PU纤维敷料表面大肠杆菌和金黄色葡萄糖球菌黏附和增殖的扫描电镜照片。
具体实施方式
下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。
如无特殊说明,下列实施例中所有原料和试剂均为市场常规的原料、试剂。
实施例1:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(3g,Mn=400)为大分子引发剂引发炔基化乙交酯单体(17.5g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.05mol%,反应温度:100℃,反应时间:24h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥48h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取5g步骤1所得侧链含炔基的生物可降解聚酯醚溶于N,N-二甲基甲酰胺并置于四口烧瓶内,通氮气,并加热至100℃;然后加入2.5g的六亚甲基二异氰酸酯,在100℃下反应4h后制得预聚体;最后,加入1.2g的扩链剂1,4-丁二醇和适量的催化剂二月桂酸二丁基锡,在100℃下继续反应2h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将6g壳寡糖(Mw=1000Da)溶于去离子水中,再加入十二烷基磺酸钠,两者摩尔比为1:2,在室温下反应2h,冷冻干燥得到烷基磺酸化的壳寡糖。(2)将上述所得烷基磺酸化壳寡糖溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:5的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖质量的1.5%,氮气保护下于0℃下反应8h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按1:10的投料比加入上述合成的固体产物和叠氮化钠,于80℃下反应6h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖溶于四氢呋喃,两者的摩尔比为1:1;然后在氮气保护下将聚合物溶液加入到含CuBr的四氢呋喃中,常温避光反应24h,得到壳寡糖修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖修饰的聚氨酯(2g)溶解于四氢呋喃和N,N-二甲基甲酰胺双溶剂中(体积比为5:5),得到质量体积浓度为12.5%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在15kV的静电压下进行纺丝,最后得到壳寡糖修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
通过表面接触角测定对静电纺丝纯聚氨酯纤维敷料(PU)和上述合成的壳寡糖修饰的聚氨酯纤维敷料(COS-PU)的亲水性进行了测试,测试结果见图1。图中数据显示,纯PU纤维膜的水接触角高达122.3±2.3°,表明纯PU纤维膜较为疏水;然而,COS-PU纤维膜的水接触角低至72.8±1.7°,说明纤维膜表面的亲水性明显得到改善。相反,以二碘甲烷为液滴相,纯PU纤维膜上的液滴接触角为8.3±1.4°,而COS-PU纤维膜表面的液滴接触角却高达46.4±1.6°。以上数据均说明采用壳寡糖修饰PU纤维膜,可以显著改善PU纤维膜表面的亲水性,有利于其更好地应用于创面敷料。
实施例2:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(5g,Mn=1000)为大分子引发剂引发炔基化乙交酯单体(22.5g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.05mol%,反应温度为120℃,反应时间为18h。反应结束后,反应粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥36h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取5g步骤1所得侧链含炔基的生物可降解聚酯醚溶于N,N-二甲基甲酰胺并置于四口烧瓶内,通氮气,并加热至120℃;然后加入3.5g的二环己基二异氰酸酯和1.0g的乙二醇,在120℃下反应5h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将5g壳寡糖(Mw=3000Da)溶于去离子水中,再加入十二烷基磺酸钠,两者摩尔比为1:5,在室温下反应4h,冷冻干燥得到烷基磺酸化的壳寡糖。(2)将上述所得烷基磺酸化壳寡糖溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:10的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖质量的2.5%,氮气保护下于10℃下反应6h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:15的投料比加入上述合成的固体产物和叠氮化钠,于80℃下反应5h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖溶于N,N-二甲基甲酰胺,两者的摩尔比为5:1;然后在氮气保护下将聚合物溶液加入到含CuBr的N,N-二甲基甲酰胺中,常温避光反应12h,得到壳寡糖修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖修饰的聚氨酯(3.5g)溶解于四氢呋喃和二甲基甲酰胺双溶剂中(体积比为3:7),得到质量体积浓度为10%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在20kV的静电压下进行纺丝,最后得到壳寡糖修饰的聚氨酯纤维敷料。
实施例3:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(5.5g,Mn=2000)为大分子引发剂引发炔基化乙交酯单体(20g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.1mol%,反应温度:120℃,反应时间:24h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥24h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取4.5g步骤1所得侧链含炔基的生物可降解聚酯醚溶于四氢呋喃并置于四口烧瓶内,通氮气,并加热至110℃;然后加入1.8g的甲苯二异氰酸酯,在120℃下反应2h后制得预聚体;最后,加入1.0g的扩链剂一缩二乙二醇,在120℃下继续反应3h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将3g壳寡糖(Mw=3000)溶于去离子水中,再加入十二烷基磺酸钠,两者摩尔比为1:8,在室温下反应1h,冷冻干燥得到烷基磺酸化的壳寡糖。(2)将上述所得烷基磺酸化壳寡糖溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:5的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖质量的1.0%,氮气保护下于20℃下反应5h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:5的投料比加入上述合成的固体产物和叠氮化钠,于100℃下反应3h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖溶于四氢呋喃,两者的摩尔比为10:1;然后在氮气保护下将聚合物溶液加入到含硫酸铜和抗坏血酸(摩尔比为1:1)的四氢呋喃中,常温避光反应6h,得到壳寡糖修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖修饰的聚氨酯(4g)溶解于N,N-二甲基甲酰胺中,得到质量体积浓度为15%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在25kV的静电压下进行纺丝,最后得到壳寡糖修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
研究了静电纺丝纯聚氨酯纤维敷料(PU)和上述合成的壳寡糖修饰的聚氨酯纤维敷料(COS-PU)对革兰氏阴性菌(大肠杆菌)和革兰氏阳性菌(金黄色葡萄糖球菌)的抑制生长能力。抑菌实验结果表明,COS-PU纤维膜抑制大肠杆菌和金黄色葡萄糖球菌的生长显著优于相应的纯PU纤维膜,表明,采用壳寡糖修饰聚氨酯,可以限制提高聚氨酯材料的抑菌性能。
表1PU和COS-PU纤维膜对大肠杆菌和金黄色葡萄球菌抑菌率
实施例4:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(5g,Mn=2000)为大分子引发剂引发炔基化丙交酯单体(12.5g)本体开环聚合,辛酸亚锡用量为炔基化丙交酯单体用量的0.15mol%,反应温度:130℃,反应时间:36h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥48h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取4g步骤1所得侧链含炔基的生物可降解聚酯醚溶于三氯甲烷并置于四口烧瓶内,通氮气,并加热至100℃;然后加入1.5g的六亚甲基二异氰酸酯,在100℃下反应4h后制得预聚体;最后,加入0.8g的扩链剂一缩二乙二醇和适量的催化剂二月桂酸二丁基锡,在100℃下继续反应3h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将5g壳寡糖(Mw=3000)溶于去离子水中,再加入一定量的十二烷基磺酸钠,两者摩尔比为1:4,在室温下反应0.5h,冷冻干燥得到烷基磺酸化的壳寡糖。(2)将上述所得烷基磺酸化壳寡糖溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:3的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖质量的2.0%,氮气保护下于0℃下反应8h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:12的投料比加入上述合成的固体产物和叠氮化钠,于90℃下反应5h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖溶于四氢呋喃,两者的摩尔比为1:2;然后在氮气保护下将聚合物溶液加入到含CuBr的四氢呋喃中,常温避光反应18h,得到壳寡糖修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖修饰的聚氨酯(3g)溶解于四氢呋喃和N,N-二甲基甲酰胺双溶剂中(体积比为2:8),得到质量体积浓度为10.0%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在15kV的静电压下进行纺丝,最后得到壳寡糖修饰的聚氨酯纤维敷料。
实施例5:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(3g,Mn=1000)为大分子引发剂引发炔基化丙交酯单体(12.5g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.15mol%,反应温度:130℃,反应时间:24h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥24h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取2.5g步骤1所得侧链含炔基的生物可降解聚酯醚溶于N,N-二甲基甲酰胺并置于四口烧瓶内,通氮气,并加热至120℃;然后加入1.2g的六亚甲基二异氰酸酯,在120℃下反应4h后制得预聚体;最后,加入0.7g的扩链剂1,4-丁二胺,在120℃下继续反应3h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将2.5g壳寡糖(Mw=5000)溶于去离子水中,再加入十二烷基磺酸钠,两者摩尔比为1:8,在室温下反应2h,冷冻干燥得到烷基磺酸化的壳寡糖。(2)将上述所得烷基磺酸化壳寡糖溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:12的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖质量的2.0%,氮气保护下于0℃下反应7h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:10的投料比加入上述合成的固体产物和叠氮化钠,于90℃下反应6h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖溶于四氢呋喃,两者的摩尔比为1:12;然后在氮气保护下将聚合物溶液加入到含硫酸铜和抗坏血酸(摩尔比为1:1)的四氢呋喃中,常温避光反应15h,得到壳寡糖修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖修饰的聚氨酯(2.5g)溶解于N,N-二甲基亚砜中,得到质量体积浓度为10%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在15kV的静电压下进行纺丝,最后得到壳寡糖修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
采用激光共聚焦观察静电纺丝纯聚氨酯纤维敷料(PU)和上述合成的壳寡糖修饰的聚氨酯纤维敷料(COS-PU)表面成纤维细胞的黏附形貌以及细胞核的分布,结果见图2。从图中可见,成纤维细胞在COS-PU纤维敷料表面的黏附面积以及细胞核的数量显著高于相应的PU纤维敷料上的成纤维细胞,这表明COS-PU纤维敷料较PU纤维敷料更有利于成纤维细胞的黏附和生长,换言之,COS-PU纤维敷料较PU敷料更有利于创面的愈合。
实施例6:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(3g,Mn=1000)为大分子引发剂引发炔基化乙交酯单体(15.5g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.05mol%,反应温度:100℃,反应时间:24h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥48h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取3g步骤1所得侧链含炔基的生物可降解聚酯醚溶于二甲基甲酰胺并置于四口烧瓶内,通氮气,并加热至100℃;然后加入1.5g的二环己基二异氰酸酯,在100℃下反应4h后制得预聚体;最后,加入0.6g的扩链剂乙二醇和适量的催化剂二月桂酸二丁基锡,在100℃下继续反应2h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将4g壳寡糖季铵盐(Mw=1150)溶于去离子水中,再加入十二烷基磺酸钠,两者摩尔比为1:3,在室温下反应4h,冷冻干燥得到烷基磺酸化的壳寡糖季铵盐。(2)将上述所得烷基磺酸化壳寡糖季铵盐溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:5的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖季铵盐质量的1.5%,氮气保护下于0℃下反应7h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:5的投料比加入上述合成的固体产物和叠氮化钠,于85℃下反应6h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖季铵盐溶于四氢呋喃,然后在氮气保护下将聚合物溶液加入到含硫酸铜和抗坏血酸(摩尔比为1:1)的四氢呋喃中,常温避光反应24h,得到壳寡糖季铵盐修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖季铵盐修饰的聚氨酯(2g)溶解于四氢呋喃和N,N-二甲基甲酰胺双溶剂中(体积比为3:7),得到质量体积浓度为12.5%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在15kV的静电压下进行纺丝,最后得到壳寡糖季铵盐修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
图3为静电纺丝聚氨酯纤维敷料(PU)和上述合成的壳寡糖季铵盐修饰的聚氨酯纤维敷料(G-COS-PU)表面成纤维细胞的黏附形貌的扫描电镜照片。从图中可见,成纤维细胞在PU纤维敷料和G-COS-PU纤维敷料表面黏附和增殖情况良好,且随着时间延长,细胞的黏附面积和数量明显增大,这表明在PU纤维敷料表面引入一定量的壳寡糖季铵盐对细胞的黏附和增殖没有显著影响。
实施例7:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(3g,Mn=2000)为大分子引发剂引发炔基化乙交酯单体(10.5g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.1mol%,反应温度:110℃,反应时间:24h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥48h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取2.5g步骤1所得侧链含炔基的生物可降解聚酯醚溶于三氯甲烷并置于四口烧瓶内,通氮气,并加热至100℃;然后加入1.5g的六亚甲基二异氰酸酯、0.8g的扩链剂1,4-丁二醇和适量的催化剂二月桂酸二丁基锡,在100℃下继续反应2h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将4g壳寡糖季铵盐(Mw=3205)溶于去离子水中,再加入十二烷基磺酸钠,两者摩尔比为1:6,在室温下反应4h,冷冻干燥得到烷基磺酸化的壳寡糖季铵盐。(2)将上述所得烷基磺酸化壳寡糖季铵盐溶于N,N-二甲基甲酰胺,加入一定量的对甲基苯磺酰氯,两者按1:8的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖季铵盐质量的2.0%,氮气保护下于10℃下反应6h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:6的投料比加入上述合成的固体产物和叠氮化钠,于90℃下反应5h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖季铵盐溶于四氢呋喃,然后在氮气保护下将聚合物溶液加入到含氯化亚铜的四氢呋喃中,常温避光反应36h,得到壳寡糖季铵盐修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖季铵盐修饰的聚氨酯(2.5g)溶解于四氢呋喃和N,N-二甲基甲酰胺双溶剂中(体积比为5:5),得到质量体积浓度为10%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在10kV的静电压下进行纺丝,最后得到壳寡糖季铵盐修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
以豚鼠为试验动物,进行材料对豚鼠皮肤的致敏实验研究。结果表明,PU和G-COS-PU试验组豚鼠试验区皮肤均无红斑、水肿,未见任何刺激反应,各时间点平均原发性刺激指数均为0(见表2)。说明G-COS-PU纤维敷料对试验豚鼠皮肤无刺激作用。试验期间各试验组动物一般状况良好(不包括皮肤局部情况),无明显异常表现。PU和G-COS-PU试验组以及阴性对照组各动物在诱导和激发期间左右两侧腹肋部及脱毛处皮肤均未见红斑、水肿等症状,激发后动物皮肤反应平均等级为0,过敏反应发生率为0%。在激发后24h及48h,阳性对照(2,4-二硝基氯苯)组各动物脱毛区皮肤均出现红斑,阳性对照组过敏反应发生率为100%(见表2)。
表2PU和G-COS-PU纤维辅料的皮肤致敏实验结果
实施例8:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(3g,Mn=400)为大分子引发剂引发炔基化丙交酯单体(12.0g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.15mol%,反应温度:130℃,反应时间:24h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥48h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取2.0g步骤1所得侧链含炔基的生物可降解聚酯醚溶于三氯甲烷并置于四口烧瓶内,通氮气,并加热至100℃;然后加入1.2g的二环己基二异氰酸酯,在90℃下反应5h后制得预聚体;最后,加入0.7g的扩链剂1,4-丁二醇和适量的催化剂二月桂酸二丁基锡,在90℃下继续反应3h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将3g壳寡糖季铵盐(Mw=2180)溶于去离子水中,再加入一定量的十二烷基磺酸钠,两者摩尔比为1:5,在室温下反应3h,冷冻干燥得到烷基磺酸化的壳寡糖季铵盐。(2)将上述所得烷基磺酸化壳寡糖季铵盐溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:9的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖季铵盐质量的2.5%,氮气保护下于10℃下反应6h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:5的投料比加入上述合成的固体产物和叠氮化钠,于90℃下反应5h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖季铵盐溶于四氢呋喃,然后在氮气保护下将聚合物溶液加入到含CuBr的四氢呋喃中,常温避光反应20h,得到壳寡糖季铵盐修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖季铵盐修饰的聚氨酯(2.0g)溶解于四氢呋喃和N,N-二甲基甲酰胺双溶剂中(体积比为4:6),得到质量体积浓度为15%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在15kV的静电压下进行纺丝,最后得到壳寡糖季铵盐修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
图4为大肠杆菌和金黄色葡萄糖球菌在静电纺丝纯聚氨酯纤维敷料(PU)和上述合成的壳寡糖季铵盐修饰的聚氨酯纤维敷料(G-COS-PU)表面的黏附和增殖情况的扫描电镜照片。图中显示,在G-COS-PU纤维敷料表面的大肠杆菌和金黄色葡萄糖球菌的数量远远小于相应的在PU纤维敷料表面大肠杆菌和金黄色葡萄糖球菌。结果表明,相比于纯PU纤维敷料,G-COS-PU纤维敷料具有更强的抑制大肠杆菌和金黄色葡萄糖球菌生长的能力,换言之,G-COS-PU纤维敷料的抑菌性能显著优于相应的纯PU纤维敷料。
实施例9:
步骤1:以辛酸亚锡为催化剂,聚乙二醇(4.5g,Mn=1000)为大分子引发剂引发炔基化乙交酯单体(16.0g)本体开环聚合,辛酸亚锡用量为炔基化乙交酯单体用量的0.15mol%,反应温度:110℃,反应时间:36h。反应结束后,粗产物用三氯甲烷溶解,无水乙醇沉淀,然后于40℃真空干燥48h,得到侧链含炔基的生物可降解聚酯醚。
步骤2:称取2.0g步骤1所得侧链含炔基的生物可降解聚酯醚溶于三氯甲烷并置于四口烧瓶内,通氮气,并加热至100℃;然后加入1.2g的甲苯二异氰酸酯,在100℃下反应8h后制得预聚体;最后,加入0.9g的扩链剂1,4-丁二醇和适量的催化剂二月桂酸二丁基锡,在100℃下继续反应4h。反应结束后,粗产物经纯化得到侧链含炔基的生物可降解聚氨酯。
步骤3:(1)将2.5g壳寡糖季铵盐(Mw=4805)溶于去离子水中,再加入一定量的十二烷基磺酸钠,两者摩尔比为1:4,在室温下反应2h,冷冻干燥得到烷基磺酸化的壳寡糖季铵盐。(2)将上述所得烷基磺酸化壳寡糖季铵盐溶于N,N-二甲基甲酰胺,加入对甲基苯磺酰氯,两者按1:6的摩尔比投料,再加入催化剂4-二氨基吡啶,催化剂用量为壳寡糖季铵盐质量的2.5%,氮气保护下于0℃下反应10h,反应产物经纯化处理。(3)在N,N-二甲基甲酰胺中,按摩尔比1:4的投料比加入上述合成的固体产物和叠氮化钠,于90℃下反应5h,反应结束后,产物经纯化和真空干燥,即得到叠氮化壳寡糖。
步骤4:将步骤2和步骤3分别所得的侧链含炔基的生物可降解聚氨酯和叠氮化壳寡糖季铵盐溶于四氢呋喃,在氮气保护下将聚合物溶液加入到含CuBr的四氢呋喃中,常温避光反应24h,得到壳寡糖季铵盐修饰的聚氨酯。
步骤5:将步骤4所得的壳寡糖季铵盐修饰的聚氨酯(2.0g)溶解于一定量的N,N-二甲基甲酰胺中,得到质量体积浓度为15%的电纺丝溶液,经过磁力搅拌、超声分散后注入电纺丝液供给装置,在20kV的静电压下进行纺丝,最后得到壳寡糖季铵盐修饰的聚氨酯纤维敷料。同样采用静电纺丝技术构建纯聚氨酯(市售,购买)纤维敷料。
以健康、初成年的白化豚鼠为试验动物,开展PU纤维敷料和G-COS-PU纤维敷料对动物创面肉芽组织生长的影响研究,结果见表3。术后第4天,各组创面清洁,表面无明显脓性分泌物附着,创基肉芽少许生长,鲜红;术后第8天,组创面清洁,表面无脓性分泌物附着,创基红润,各组创面清洁,肉芽大量生长,鲜红颗粒状,触之易出血。表3中各组肉芽组织生成量数据显示,G-COS-PU纤维敷料组上4天和8天肉芽组织生成量均高于相应的PU纤维敷料组,表明G-COS-PU纤维敷料相比于PU纤维敷料更有利于创面的修复和愈合。
表3PU和G-COS-PU纤维辅料对动物创面肉芽组织生长的影响
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!