一种基于酰基自由基的产生制备双取代氧化吲哚的方法与流程
本发明涉及一种酰基自由基产生方法,具体涉及一种采用催化剂制得酰基自由基,并用于制备双取代氧化吲哚。
背景技术:
酰基自由基作为一类有用的结构单元,可以用于杂环化合物中功能化碳碳键的构建,但与烷基和烯丙基自由基相比,酰基自由基由于具有强亲核性和严苛的制备条件使其发展受到了很大限制。
目前,简洁、高效的酰基自由基产生方法受到了化学家的广泛关注。醛自发氧化生成酰基自由基的方法是一类具有高原子经济性的酰基自由基产生方法,该方法不需要额外加入其他试剂,只需通过醛自发反应便可得到酰基自由基,但该方法反应效率低,所需时间较长,一般需要3-6天。光催化反应是一类高效、绿色、可持续的催化反应模式,与常规的酰基自由基产生方法相比,光催化反应可以凭借其特殊的反应机理高效产生酰基自由基。光催化产生酰基自由基的方法主要有:α-酮酸的脱羧化、酸酐的脱羧化和羧酸的脱羧插羰化。在α-酮酸的脱羧化、酸酐的脱羧化反应中,都需要脱除一分子二氧化碳才能得到酰基自由基,而且部分底物需要额外合成,限制了反应在实际应用中的价值。在羧酸的脱羧插羰化反应中,羧酸无法直接形成酰基自由基,必须在脱除一分子二氧化碳后,向反应体系中加入一分子一氧化碳才能实现酰基自由基的产生,缺乏原子经济性。除此之外,α-酮酸的脱羧化、酸酐的脱羧化和羧酸的脱羧插羰化在反应中多采用二甲基亚砜、N,N-二甲基甲酰胺等高沸点溶剂,这一类溶剂必须在反应结束后经过反复水洗才能除去,使反应的后处理变得复杂。
技术实现要素:
本发明提供了一种基于酰基自由基的产生制备双取代氧化吲哚的方法,采用酰氯一步得到酰基自由基,提高了原子的经济型。
本发明的目的通过以下技术方案来具体实现的:
一种基于酰基自由基的产生制备双取代氧化吲哚的方法,采用催化剂制得酰基自由基,并用于制备双取代氧化吲哚。
进一步的,具体包括如下步骤:
(1)将催化剂、N-取代酰胺和溶剂加入反应器,液氮冷却脱气后充入氩气,反复操作三次保证反应体系处于惰性气体环境;
(2)将碱、酰氯加入步骤(1)氩气保护的反应器中,在光照射下反应;
(3)待步骤(2)反应完后,蒸干溶剂,加水和乙酸乙酯对剩余物进行萃取,经分离处理后得到双取代氧化吲哚。
更进一步的,所述步骤(2)中的反应在10~35℃进行,反应时间为6~18h,光照射的光源为25W 蓝光LED光带。
更进一步的,所述步骤(1)中的催化剂为光催化剂,选自包括fac-Ir(PPy)3、[Ir(ppy)2(dtbbpy)]PF6、[Ir{dF(CF3)ppy}2(dtbbpy)]PF6、Ru(phen)3Cl2、Ru(bpy)3Cl2•6H2O、Cu(dap)2Cl、Eosin Y中的一种,催化剂的用量占N-取代酰胺摩尔数的1~2%。
更进一步的,所述步骤(1)中N-取代酰胺的用量为0.05~0.2mmol,具有通式:
式(A)中R1、R2、R3、R4、R5基团可以是相同或不同的,包括氢原子、卤素原子、烷基、烷氧基、芳基,所述卤素原子优选包括氟原子、氯原子、溴原子,所述烷基优选包括甲基、乙基、叔丁基,所述烷氧基优选包括甲氧基、乙氧基,所述芳基优选包括苯基;
式(A)中R6基团包括烷基、芳基和酰基,所述烷基优选包括甲基、乙基,所述芳基优选包括苄基、苯基,所述酰基优选包括乙酰基。
更进一步的,所述步骤(1)中溶剂的用量为2~6mL,包括乙腈、N,N-二甲基甲酰胺、丙酮、乙酸乙酯、四氢呋喃、二氯甲烷、1,2-二氯乙烷、三氯甲烷、苯、甲苯、二甲苯中的一种,优选为乙腈、丙酮、乙酸乙酯和1,2-二氯乙烷。
更进一步的,所述步骤(2)中碱的用量为0.1~0.3mmol,包括甲基吡啶、2,6-二甲基吡啶、三乙胺、N,N-二异丙基乙胺、三乙烯二胺、4-二甲氨基吡啶、1,8-二氮杂二环十一碳-7-烯、磷酸氢二钠、碳酸钠、碳酸铯中的一种,优选为甲基吡啶、2,6-二甲基吡啶、三乙胺、磷酸氢二钠和碳酸钠。
更进一步的,所述步骤(2)中酰氯的用量为0.05~0.2mmol,具有通式:
或
式(B)中R7、R8、R9、R10、R11基团可以是相同或不同的,包括氢原子、卤素原子、烷基、烷氧基和芳基,所述卤素原子优选包括氟原子、氯原子、溴原子,所述烷基优选包括甲基、三氟甲基、乙基、异丙基、叔丁基,所述烷氧基优选包括甲氧基、乙氧基,所述芳基优选包括苯基;
式(C)中R12、R13、R14基团可以是相同或不同的,包括氢原子、卤素原子、烷基和烷氧基,X为硫原子或氧原子,所述卤素原子优选包括氟原子、氯原子、溴原子,所述烷基优选包括甲基、三氟甲基、乙基、异丙基、叔丁基,所述烷氧基优选包括甲氧基、乙氧基。
更进一步的,所述步骤(3)中的双取代氧化吲哚具有通式:
或
式(D)和式(E)中R2、R3、R4、R5基团可以是相同或不同的,包括氢原子、卤素原子、烷基、烷氧基、芳基,所述卤素原子优选包括氟原子、氯原子、溴原子,所述烷基优选包括甲基、乙基、叔丁基,所述烷氧基优选包括甲氧基、乙氧基,所述芳基优选包括苯基;
式(D)和式(E)中R6基团包括烷基、芳基和酰基,所述烷基优选包括甲基、乙基,所述芳基优选包括苄基、苯基,所述酰基优选包括乙酰基;
式(D)中R7、R8、R9、R10、R11基团可以是相同或不同的,包括氢原子、卤素原子、烷基、烷氧基和芳基,所述卤素原子优选包括氟原子、氯原子、溴原子,所述烷基优选包括甲基、三氟甲基、乙基、异丙基、叔丁基,所述烷氧基优选包括甲氧基、乙氧基,所述芳基优选包括苯基;
式(E)中R12、R13、R14基团可以是相同或不同的,包括氢原子、卤素原子、烷基和烷氧基,X为硫原子或氧原子,所述卤素原子优选包括氟原子、氯原子、溴原子,所述烷基优选包括甲基、三氟甲基、乙基、异丙基、叔丁基,所述烷氧基优选包括甲氧基、乙氧基。
本发明依据的技术原理为:
如上式所示,本发明中光催化剂在光激发下跃迁至激发态,进行单电子转移过程还原酰氯,酰氯失去氯离子形成酰基自由基。酰基自由基与N-取代酰胺发生自由基环化反应,生成双取代氧化吲哚。
本发明的有益效果为:
(1)本发明所提供的产生酰基自由基的方法避免了传统光催化自由基脱羧过程和当量二氧化碳气体的产生,同时也避免了额外一氧化碳引入过程,采用酰氯一步得到酰基自由基,简化了反应历程,提高了原子经济性;
(2)本发明所用酰氯为廉价易得的化工原料,避免了额外合成酰基自由基前体,而且酰氯易溶于常见有机溶剂,反应过程中不用加入N,N-二甲基甲酰胺(DMF)和二甲基亚砜(DMSO)等高沸点溶剂,避免了此类溶剂后处理复杂的问题,提升了该方法的实际应用价值;
(3)本发明具有广泛的应用价值,可以经济、高效的合成双取代氧化吲哚,该类化合物是一种用途广泛的医药化工中间体,有很高的科研和应用价值。
附图说明
图1为本发明酰基自由基验证中苯甲酰氯的电位图,横坐标为电位值(V),纵坐标为电流值(A);图2为本发明实施例1的1H NMR谱图;图3为本发明实施例1的13C NMR谱图;图4为本发明实施例2的1H NMR谱图;图5为本发明实施例2的13C NMR谱图;图6为本发明实施例3的1H NMR谱图;图7为本发明实施例3的13C NMR谱图;图8为本发明实施例4的1H NMR谱图;图9为本发明实施例4的13C NMR谱图;图10为本发明实施例5的1H NMR谱图;图11为本发明实施例5的13C NMR谱图;图12为本发明实施例6的1H NMR谱图;图13为本发明实施例6的13C NMR谱图;图14为本发明实施例7的1H NMR谱图;图15为本发明实施例7的13C NMR谱图;图16为本发明实施例8的1H NMR谱图;图17为本发明实施例8的13C NMR谱图。
具体实施方式
以下对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
本发明实施例中原料的来源和规格如下:
光催化剂 阿拉丁试剂
酰氯以及其他分析纯试剂 安耐吉试剂
柱层析硅胶(200~300目) 青岛海洋化工有限公司
反应用无水无氧溶剂采用Innovative TechnologyPureSolv MD-5型溶剂纯化系统制得。
光催化反应在蓝光反应器中进行,光源为25W蓝色LED光带。
N-取代酰胺由以下技术路线制得:
其中方法1适用于芳环上带有取代基的苯胺,方法2适用于氮原子上带有取代基的苯胺。
具体制备过程参考文献Organic Letters 2015, 17(9): 2142−2145。制备的N-取代酰胺具有如下通式:
酰氯具有如下通式:
分析方法
(1)酰基自由基和技术原理验证由循环伏安法测定,方法如下:采用CHI660E电化学工作站,所添加电解质为Bu4NBF4,所用电极为Ag/AgCl。
(2)双取代氧化吲哚结构由(1H NMR和13C NMR)测定,方法如下:采用Bruker-400型和VarianMercury-300型核磁共振仪测定,CDCl3为溶剂,四甲基硅烷(TMS)为内标。
酰基自由基验证
以Ag/AgCl为电极,测得苯甲酰氯(R7=R8=R9=R10=R11=H)的还原电位为-0.972 V vs Ag/AgCl,Ag/AgCl电极所测得的还原电位比饱和干汞电极测得的还原电位要高45.9 mV,所以苯甲酰氯的还原电位也可以表示为-1.018 V vs SCE。激发态光催化剂Ir(ppy)3的还原电位为E1/2IV/*III = -1.73 V,表明该催化剂可以还原苯甲酰氯产生酰基自由基。
实施例1
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R3=R4=R5=H,R6=Me)17.5mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R7=R8=R9=R10=R11=H)28mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚26.5mg,产率95%。
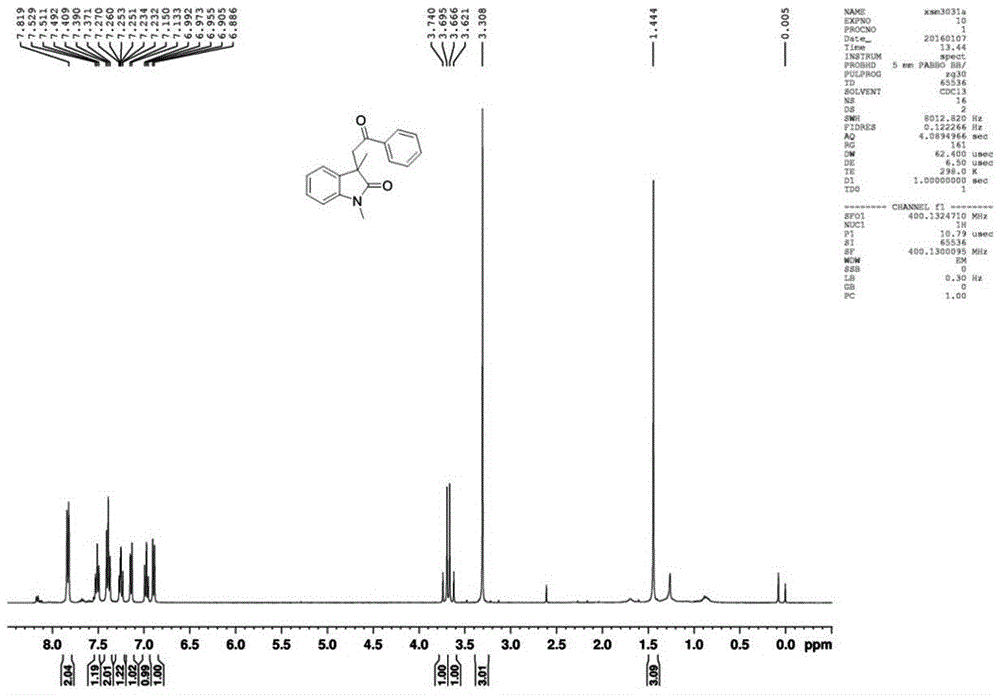
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.31处有信号,与氧化吲哚季碳中心相连的甲基在1.44处有信号,与羰基相连的亚甲基分别在3.65和3.72处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.5处有信号,与氧化吲哚季碳中心相连的甲基在25.1处有信号,与羰基相连的亚甲基在46.2处有信号。
实施例2
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R3=R4=R5=H,R6=Me)17.5mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入甲基吡啶23.3mg(0.25mmol)和酰氯(R7=Me,R8=R9=R10=R11=H)30.8mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚16.4mg,产率56%。
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.35处有信号,与氧化吲哚季碳中心相连的甲基在1.76处有信号,与羰基相连的亚甲基分别在3.65和3.72处有信号,酰氯苯环上甲基取代基(R7)在2.70处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.5处有信号,与氧化吲哚季碳中心相连的甲基在25.1处有信号,与羰基相连的亚甲基在46.2处有信号,酰氯苯环上甲基取代基(R7)在20.3处有信号。
实施例3
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R3=R4=R5=H,R6=Me)17.5mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R8=Cl,R7=R9=R10=R11=H)34.8mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚20.7mg,产率66%。
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.31处有信号,与氧化吲哚季碳中心相连的甲基在1.44处有信号,与羰基相连的亚甲基分别在3.50和3.60处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.4处有信号,与氧化吲哚季碳中心相连的甲基在24.8处有信号,与羰基相连的亚甲基在46.1处有信号,酰氯苯环上氯取代基(R8)在20.3处有信号。
实施例4
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R3=R4=R5=H,R6=Me)17.5mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R8=R10=Me,R7=R9=R11=H)33.7mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚27.1mg,产率88%。
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.31处有信号,与氧化吲哚季碳中心相连的甲基在1.43处有信号,与羰基相连的亚甲基分别在3.60和3.70处有信号,酰氯苯环上取代基(R8、R10)在2.31处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.4处有信号,与氧化吲哚季碳中心相连的甲基在24.8处有信号,与羰基相连的亚甲基在46.1处有信号,酰氯苯环上甲基取代基(R8、R10)在21.1处有信号。
实施例5
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R5=R4=H,R3= R6=Me)18.9mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R7=R8=R9=R10=R11=H)28mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚29.3mg,产率87%。
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.28处有信号,与氧化吲哚季碳中心相连的甲基在1.42处有信号,与羰基相连的亚甲基分别在3.63和3.69处有信号,与N-取代酰胺苯环相连的甲基(R6)在2.27处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.5处有信号,与氧化吲哚季碳中心相连的甲基在25.0处有信号,与羰基相连的亚甲基在46.5处有信号,与N-取代酰胺苯环相连的甲基(R6)在21.1处有信号。
实施例6
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R5=R4=H,R3=Br,R6=Me)25.4mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R7=R8=R9=R10=R11=H)28mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚29.4mg,产率82%。
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.30处有信号,与氧化吲哚季碳中心相连的甲基在1.42处有信号,与羰基相连的亚甲基在3.69处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.5处有信号,与氧化吲哚季碳中心相连的甲基在24.9处有信号,与羰基相连的亚甲基在46.1处有信号。
实施例7
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R5=R4=H,R3=OMe,R6=Me)20.5mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R7=R8=R9=R10=R11=H)28mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚26.9mg,产率87%。
测定:在双取代氧化吲哚1H NMR谱图中,与氮原子相连的甲基在3.29处有信号,与氧化吲哚季碳中心相连的甲基在1.43处有信号,与羰基相连的亚甲基分别在3.64和3.69处有信号,与N-取代酰胺苯环相连的甲氧基(R3)在3.73处有信号。在双取代氧化吲哚13C NMR谱图中,氮原子相连的甲基在26.6处有信号,与氧化吲哚季碳中心相连的甲基在24.8处有信号,与羰基相连的亚甲基在45.4处有信号,与N-取代酰胺苯环相连的甲氧基(R3)在46.1处有信号。
实施例8
双取代氧化吲哚的制备:在氩气保护下,称取2mol%光催化剂fac-Ir(PPy) 1.31mg和N-取代酰胺(R1=R2=R3=R4=R5=H,R6=Bn)25.1mg(0.11mmol)加入反应管中,同时加入磁子,塞紧瓶塞后用注射器加入4mL新制无水乙腈,溶液用液氮冷却,抽真空,充氩气,溶解,反复操作三次,保证反应体系处于惰性气体环境。用微量进样器分别加入2,6-二甲基吡啶26.9mg(0.25mmol)和酰氯(R7=R8=R9=R10=R11=H)28mg(0.20mmol),将反应体系完全密封后置于蓝光光反应器,保持反应温度为20℃。
反应12小时后,蒸干反应体系内溶剂,加蒸馏水和乙酸乙酯对剩余物进行萃取,所得产物用柱层析色谱分离纯化,所用洗脱剂为正己烷和乙酸乙酯,称重后得双取代氧化吲哚33.4mg,产率94%。
测定:在双取代氧化吲哚1H NMR谱图中与氧化吲哚季碳中心相连的甲基在1.43处有信号,与羰基相连的亚甲基分别在3.72和3.78处有信号,与N-取代酰胺氮原子相连苄基(R6)分别在5.09和6.73处有信号。在双取代氧化吲哚13C NMR谱图中,与氧化吲哚季碳中心相连的甲基在25.5处有信号,与羰基相连的亚甲基在45.4处有信号,与N-取代酰胺氮原子相连苄基(R6)在45.8处有信号。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!