一种制备对乙烯基苯酚的方法与流程
本发明涉及一种制备对乙烯基苯酚的生物催化方法。
背景技术:
乙烯基苯酚衍生物在聚合物化学中被用作有用组分,并且例如能够在制造化学和生物传感器时用于组装电介质层。卤化衍生物例如被应用于制造阻燃剂以及查尔酮,一类已知的具有广泛生物活性的有机化合物。
将非活化的酚类选择性对位乙烯基化为对-或4-乙烯基苯酚并不已知。因此,在使用锡催化剂的情况下,非活化芳烃的直接乙烯基化生成选择性的邻位衍生物(yamaguchi等人,j.am.chem.soc.117,1151-1152(1995)),而用路易斯酸(例如gacl3)催化提供邻位和对位区域异构体的混合物(yamaguchi等人,angew.chem.int.ed.36,1313-1315(1997))。然而,在苯酚衍生物上并不实施这两种方法。更多的是借助施蒂勒反应(stille-kupplung)在使用相应的对溴苯酚和非常毒性的乙烯基锡试剂来人工合成对乙烯基苯酚(littke等人,j.am.chem.soc.124,6343-6348(2002))。
替代地,对乙烯基苯酚能够由相应的4-羟基苯甲醛借助赫克反应在鏻盐催化作用下(chen等人,tetrahedron69,653-657(2013))或者通过诺文葛尔缩合反应(knoevenagel-kondensation)在根据多布纳的变体中借助仲胺的催化作用在微波辐照(sinha等人,tetrahedron63,960-965(2007))下制备而成。
相比之下,生物催化方法局限于分别使用离子液体(sharma等人,adv.synth.catal.350,2910-2920(2008))、酚酸脱羧酶(pad)(wuensch等人,org.lett.14,1974-1977(2012))、或者特定的对羟基肉桂酸或对香豆酸脱羧酶(phca-dc或pca-dc或简称为pdc)例如来自植物乳杆菌的pdc(rodriguez等人,proteins78,1662-1676(2010)和jung等人,appl.microbiol.biotechnol.97,1501(2013))使对香豆酸(4-羟基肉桂酸)脱羧,任选地在两相体系中(ben-bassat等人,org.processres.dev.11,278-285(2007)):
然而,为此通常必须由相应的醛来合成相应的肉桂酸,这是昂贵的。也报道过使用共同表达真菌苯基氨裂解酶(pal)和细菌对羟基肉桂酸脱羧酶(pdc)的基因工程细菌,通过消除酪氨酸中的氨,从葡萄糖来细菌生产对乙烯基苯酚。然而生产率在此极低(0.4g/l),反应局限于该酶底物,诸如苯基丙氨酸(0.5g/l)和肉桂酸的副产物的量相当大(qi等人,metabolicengin.9,268-276(2007))。
另外,基于苯酚,酪氨酸衍生物的生物催化人工合成方法是已知的:
发明人工作团队的出版物,kroutil等人,adv.synth.catal.352,731-736(2010)公开了一系列苯酚衍生物的此种反应,其中取代基f、cl、br、ch3和och3在苯酚的2或3位置上,并且是在使用了弗氏柠檬酸杆菌(citrobacterfreundii)的酪氨酸苯酚裂解酶(tpl)的情况下进行的。
借助酪氨酸氨裂解酶(tal)或苯基氨裂解酶(pal)从酪氨酸中酶促地催化消除氨以获得肉桂酸同样是已知的:
参见kyndt等人,febsletters512,240(2002),以及louie等人,chem.biol.13(12),1327-1338(2006)。然而,至今未曾报道此反应用于酪氨酸的衍生物。
在这样的背景下,本发明的目的在于开发更好的生物催化方法用于制备对乙烯基苯酚,该方法能够实现对乙烯基苯酚的迅速且经济实惠的生产。
技术实现要素:
本发明通过提供一种用于制备对乙烯基苯酚的生物催化方法来实现此目的,该方法包括依据下述反应流程图的三步一锅反应:
其中,
a)以自身已知的方式将可被取代的苯酚1借助酪氨酸酚裂解酶(tpl)的催化作用并且在铵离子存在的情况下与丙酮酸(bts)结合生成可被取代的酪氨酸2,
b)为了获得可被取代的对香豆酸3,以自身已知的方式借助酪氨酸氨裂解酶(tal)或苯基氨裂解酶(pal)的催化作用从酪氨酸2消除氨,和
c)为了获得期望的可被取代的对乙烯基苯酚4,以自身已知的方式借助酚酸脱羧酶(pad)的催化作用使对香豆酸3脱羧;
d)其中将产生的co2排出反应体系,以使所有三个反应级的化学平衡都向着产物的方向移动。
本发明在此基于三个自身已知的单一反应步骤的结合以及出乎意料的认知,即不仅所有三个反应能够基本上同时在一个一锅反应里进行,而不至于在一定程度上相互抑制,而且完全能够达到超过90%的转化率——即使是以被取代的苯酚作为起始底物。这一点并不是所希望的,主要是基于以下事实,即不仅反应物而且全部产物在形式上都是苯酚,这样的话在三个单一反应中的相互抑制作用可能性就较高。
通过将三个反应步骤实施为一锅反应,并且因为将脱羧时在步骤c)中产生的二氧化碳排出体系而使得总反应的化学平衡移动,发明人在优化了反应条件之后甚至成功地实现了总转化率(通过全部三级计算所得)达到全部高于97%,如后面的实施例所证实的那样,这是完全没有预料到的。
因此,本发明提供了一种方法,该方法以较高的纯度实现了从苯酚向对乙烯基苯酚的可选择性的、非常快速的并且几乎完全的转化,这是以传统的有机化学方式至今仍不可能实现的。
如发明人通过变化ph值所确定的那样,根据本发明的一锅反应在优选的实施方式中在ph值为8至9时,优选ph值为8来实施。极好的整体转化率在这方面也是非常令人吃惊,因为两个第一反应步骤的理想ph值分别都高于10,而脱羧反应在ph为酸性至中性时比在碱性氛围里进行得明显更快、更完全,而且对于单个反应来说自ph值超过9就几乎观察不到任何转化,正如稍后示例中再次证实的那样。单一反应的结果与开始所引用的专业文献(之前的rodriguez等人;之前的jung等人,)相符,虽然jung等人(之前的)公开了在ph值为5.8时他们所使用的pad的ph最优值,但是所引用的专业文献里却例如在ph值为6.5或7.0时借助pad来进行生物催化脱羧。
但由于这个原因,在ph为8时根据本发明的一锅反应也被证实为理想的,因为在该ph值下可以使用无毒的kpi缓冲液(磷酸钾)作为水性溶液,而当ph为9时则必须使用例如磺酸盐类(例如ches缓冲液:n-环己基-2-氨基乙磺酸)的更毒的试剂。
在另一优选实施方式中,在b)步骤中使用酪氨酸氨裂解酶(tal)作为催化剂,因为发明人用研究过的苯基氨裂解酶(pal)几乎不能发现转化。优选地,以包含重组酶的全细胞形式加入酪氨酸氨裂解酶(tal),因为它们在根据本发明的方法中表现出了足够高的活性,并且因此能够避免昂贵的且有时很大浪费地从细胞培养物中分离酶。
同样的情况也适用于根据本发明优选在步骤c)中作为催化剂使用的阿魏酸脱羧酶(fad),它在此特别优选地也以包含重组酶的全细胞的形式加入。
在其他优选的实施方式中,除了带有相应ph值的水性缓冲系统外还使用了与水不混溶的共溶剂,优选的是乙醚,特别优选的是相对于水性缓冲体系而言5%的乙醚,以进一步提高整个反应的转化率。
起始苯酚1上的取代模式并非有特殊限定,只要对位并未被占据,并且取代对步骤a)至c)的酶反应没有负面影响。如后面示例中所示出的那样,本发明不仅通过降低芳香环电子密度的取代基如f、cl和br起作用,而且也通过提高电子密度的取代基如烷基或烷氧基起作用。为了不妨碍特别是在步骤a)中与各个酶的配位,取代基团的大小在此不应该超过特定尺寸,这就是为何优选含有不多于20个碳原子的烃基,更加优选含有不超过10个或者不超过6个碳原子的烃基的原因。特别优选地,在苯酚1的邻位或间位上的取代基选自卤素以及c1-6烷基和c1-6烷氧基,例如f、cl、br或(o)ch3。
附图说明
随后将参考附图对本发明做进一步说明,详见如下:
图1:在不同ph值下步骤a)中的转化率;
图2:在不同ph值下步骤b)中的转化率;
图3:在不同ph值下步骤c)中的转化率;
图4:在不同ph值下总反应随时间的进程;
图5:ph为7时反应物及产物的量;
图6:ph为10时反应物及产物的量;
图7:使用不同共溶剂时总反应的转化率;和
图8:在理想反应条件下反应物及产物的量。
实施例
下面为本发明的实施例,这些实施例是为了说明,但并不可理解为是对保护范围的限定。
一般方法
从不同的商业渠道获得化学试剂且使用时无需进一步纯化。通过在开放毛细管中的样品来确定熔点且不修正。使用布鲁克光谱仪获得1h-和13c-核磁共振谱图(1h:300.13mhz;13c:75.5mhz)。以ppm给出化学位移,以赫兹(hz)给出偶合常数。以不同的波长在带有紫外探测器的岛津色谱仪上使用鲁娜c18色谱柱(
准备样品:将mecn/h2o溶液(1ml,1∶1)与0.1%tfa注入反应混合物的等分(1ml)当中。通过离心分离出蛋白质,通过vivaspin聚醚砜半透膜过滤器过滤溶液。借助hplc在下面给出的条件下分析所得溶液。由各自峰面积计算出化合物的相对含量。色谱柱:鲁娜-c18,5μm;流量:1ml/min;温度:30℃;梯度:22分钟内从100%h2o(0.1%tfa)到100%mecn(0.1%tfa)。波长:280nm。
人工合成实施例1至3:制备酶制剂
人工合成实施例1
由弗氏柠檬酸杆菌m379v(citrobacterfreundiim379v)制备作为无细胞提取物的tpl
包含有m379v-tpl-质体的大肠杆菌克隆在lb培养基中繁殖,通过在5个11的锥形瓶中消毒如下成分的溶液(11)来制造该lb培养基:胰蛋白胨(10g/l),nacl(5g/l)和酵母抽提物(5g/l)。通过接种100ml含有氨苄青霉素(100mg/l)的lb培养基制备预培养物。在120转/分钟和37℃下过夜摇动预培养物。之后在包含有氨苄青霉素(100mg/l)的烧瓶中接种预培养物,这得到0.05的起始od600。培养物接下来在120转/分钟和30℃下摇动直至达到0.4至0.6的od600。用iptg(0.5mm,最终浓度)诱导蛋白质表达,并且在20℃和120转/分钟时摇动培养物2小时。最后,通过离心分离(8000转/分钟,20分钟)获得细胞,并以磷酸钾缓冲液(10mm,ph7)清洗,重新悬浮在kpi缓冲液(50mm,180mmnh4cl,0.04mmplp,ph8)中,并借助超声波处理(40%振幅,1秒脉冲开,2秒脉冲关,5分钟)来破坏。通过离心分离(15000转/分钟,15分钟)获得混合物,浮在上层的物质在液氮中快速冷冻并冻干。在4℃下保存冻干的无细胞提取物并且按原样地应用到反应中。
人工合成实施例2
由球形红细菌(rhodobactersphaeroides)制备作为全细胞催化剂的tal
包含有tal-质体的大肠杆菌克隆在lb培养基中繁殖,通过在5个11的锥形瓶中消毒如下成分的溶液(11)来制造该lb培养基:胰蛋白胨(10g/l),nacl(5g/l)和酵母抽提物(5g/l)。通过接种含有卡那霉素(50mg/l)的100mllb培养基来制备预培养物。在120转/分钟和37℃下过夜摇动预培养物。之后在包含有卡那霉素(50mg/l)的烧瓶中接种预培养物,这得到0.05的起始od600。培养物接下来在120转/分钟和37℃下摇动直至达到0.5至0.7的od600。用iptg(0.5mm,最终浓度)诱导蛋白质表达,并且在20℃和120转/分钟时摇动培养物24小时。最后,通过离心分离(8000转/分钟,20分钟)获得细胞,并以磷酸钾缓冲液(10mm,ph8)清洗,在液氮中快速冷冻并冻干。在4℃下保存冻干的细胞并且按原样地应用到反应中。
人工合成实施例3
由肠杆菌属(enterobactersp.)制备作为大肠杆菌全细胞催化剂的fad
包含有fad-质体的大肠杆菌克隆在lb培养基中繁殖,通过在5个1l的锥形瓶中消毒如下成分的溶液(11)来制造该lb培养基:胰蛋白胨(10g/l),nacl(5g/l)和酵母抽提物(5g/l)。通过接种含有卡那霉素(50mg/l)的100mllb培养基来制造预培养物。在120转/分钟和37℃下过夜摇动预培养物。之后在包含有卡那霉素(50mg/l)的烧瓶中接种预培养物,这得到0.05的起始od600。培养物接下来在120转/分钟和37℃下摇动直至达到0.5至0.7的od600。用iptg(0.5mm,最终浓度)诱导蛋白质表达,并且在20℃和120转/分钟时摇动培养物24小时。最后,通过离心分离(8000转/分钟,20分钟)获得细胞,并以磷酸钾缓冲液(10mm,ph8)清洗,在液氮中快速冷冻并冻干。在4℃下保存冻干的细胞并且按原样地应用到反应中。
参考实施例1至3:活性测量
参考实施例1
tpl的活性
为了更好的比较酶活性,通过借助hplc测量起始反应速度来确定tpl在根据步骤a)的苯酚1、丙酮酸
参考实施例2
tal的活性
为了更好地比较酶活性,通过借助hplc测量起始反应速率来确定tal在根据步骤b)的酪氨酸2向肉桂酸衍生物3的转化中的活性。活性单元被定义为在以下条件时每分钟催化合成1μmol肉桂酸3的催化剂用量:30℃,kpi缓冲剂50mm,ph8,850转/分钟。测试混合物包含有l-3-氯酪氨酸(2b,5mm),nh4cl(180mm)和含有过度表达tal(5mg)的来自人工合成实施例2的大肠杆菌全细胞。通过加入酶来启动反应,并在5至20分钟之间确定转化率。所有测量至少一式三份地进行。确定的反应活性对应于9.2mu每毫克的大肠杆菌全细胞。
参考实施例3
fad的活性
为了更好地比较酶活性,通过借助hplc测量起始反应速率来确定fad在根据步骤c)的肉桂酸衍生物3脱羧生成乙烯基苯酚4中的活性。活性单元被定义为在以下条件时每分钟催化合成1μmol乙烯基苯酚4的催化剂用量:30℃,kpi缓冲剂50mm,ph8,850转/分钟。测试混合物包含有3-氯-4-羟基肉桂酸(3-氯香豆酸,3b,5mm),nh4cl(180mm)和含有过度表达fad(0.1mg)的来自人工合成实施例3的大肠杆菌全细胞。通过加入酶来启动反应,并在1至5分钟之间确定转化率。所有测量至少一式三份地进行。确定的反应活性对应于2.1u每毫克的大肠杆菌全细胞。
参考实施例4至6:确定最优ph值
参考实施例4
tpl的最优ph值
使用来自人工合成实施例1的冻干的无细胞提取物的在2-氯苯酚1b(23mm)、丙酮酸盐(46mm)和nh4+(180mm)之间的参考实施例1的tpl催化的酶促反应在加入0.4mm磷酸吡哆醛作为辅酶的情况下在变化的ph值下被重复并被进行22小时。图1示出了ph值6至10的结果,由此得出,在强碱性ph下得到l-3-氯酪氨酸2b的最佳转化率,因为不带电的氨在这一范围内能够参与反应。有趣的是,酶自身在强碱性ph范围内维持稳定,尤其是在ph值为10时甚至得到了最佳的转化率,因此,最优值可能还在此之上。
参考实施例5
tal的最优ph值
使用来自人工合成实施例2的全细胞制剂的在3-氯酪氨酸2b(10mm)和nh4+(180mm)之间的参考实施例2的tal催化的酶促反应在加入0.4mm磷酸吡哆醛(plp)作为辅酶的情况下在变化的ph值下也被重复并且被进行6小时。图2再次示出了ph值6至10的结果,由此再次得出,优选高于ph10的强碱性ph范围。在此情况下,反应的理想值肯定是高于ph10,如通过经过测量点的理论最佳拟合曲线的外推所表明的。
参考实施例6
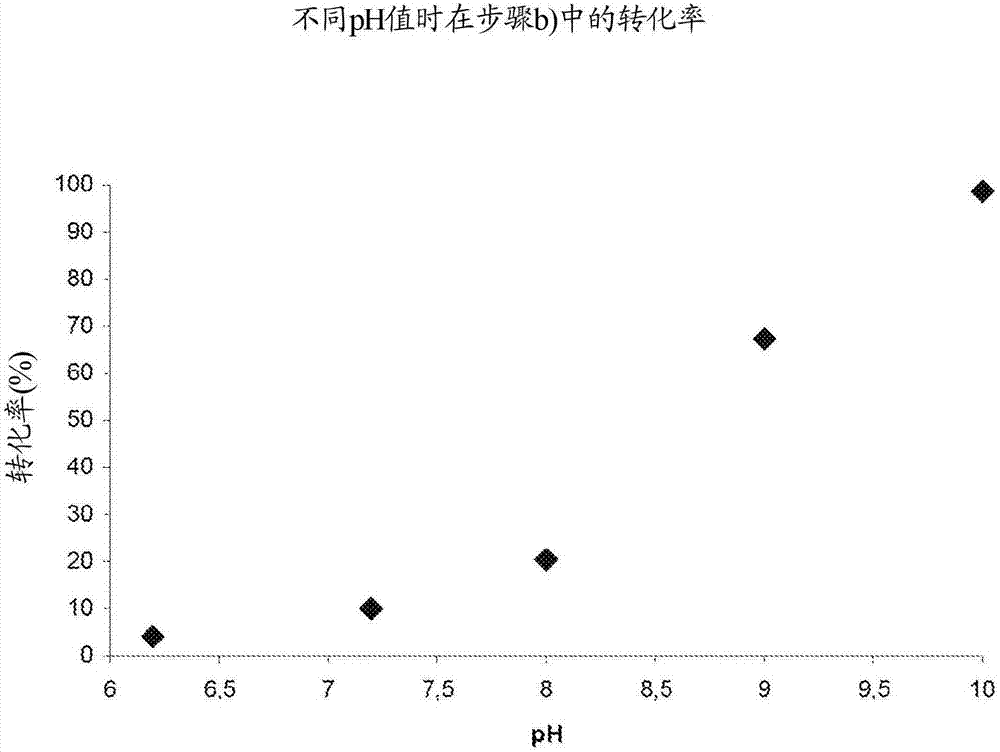
fad的最优ph值
参考实施例3的fad催化的酶促反应,即,使用来自人工合成实施例3的全细胞制剂使3-氯香豆酸3b(10mm)脱羧得到4-乙烯基苯酚4b在加入0.4mm磷酸吡哆醛(plp)作为辅酶的情况下在变化的ph值下被重复并且被进行1小时。图3再次示出了ph值6至10的结果,由此得出,直至ph8能够得到比得上在中性范围内的转化率的转化率,然而ph为9时几乎得不到转化,并且ph为10时实际上不再转化。在此情况下,反应的理想值肯定是处于酸性ph范围中,如通过理论最佳拟合曲线的外推所表明的,并且如本领域技术人员对于脱羧反应所预期的。
实施例1至4
不同ph值时,步骤a)至c)的一锅反应
使用人工合成实施例1至3中的三种酶,以如下条件进行使用2-氟苯酚1a(r=f)的三步一锅反应:1a(23mm);bts(丙酮酸盐)(46mm);tpl(4mg);tal(20mg);fad(5mg);水性缓冲剂(50mm):kpi用于ph7-8,ches用于ph9-10;时间:6小时;20℃;以850转/分钟摇动。ph值在7至10之间变化,以时间间隔提取样品,并借助hplc分析2-氟-4-乙烯基苯酚4a(r=f)的含量。
图4示出了这些反应的结果。结果表明ph为7(实施例1;大约30%)时或ph为10(实施例4;大约45%)时的转化率明显低于ph为8(实施例2;大约85%)时和ph为9(实施例3;大约80%)时,对它们来说几乎得到相同的转化率,这一方面可以归因于tpl和tal两种酶在ph为7时(参看图1和2)活性较低,另一方面还能够归因于fad在ph为10(参看图3)时几乎不起作用。考虑到后一种情况令人吃惊的是,在ph为10时却比在ph为7时可得到明显更好的转化率。
为了研究这一现象,在ph为7和ph为10时重复实施例1和4,并借助hplc在此时间间隔内确定反应物1a和产物2a至4a的量。
图5和6示出了对于苯酚1a(▲)、酪氨酸2a(◆)、肉桂酸3a(■)和所期望得到的乙烯基苯酚4a(x)的实验结果。这证实了上面的推测,因为这里示出了反应物1a的量在ph为7时仅仅非常缓慢地减少,并且在6小时后仍存有40%以上的初始苯酚,而实际上在反应混合物中始终没有获得肉桂酸。为此,当ph为7时步骤a)是决定速度的部分反应,而以最快的速度进行步骤c)的脱羧。相反,当ph为10时步骤a)的c-c偶联明显更快,从而在6小时后起始产物1a下降到原始量的10%。而同时肉桂酸3a连续地在反应混合物中聚集,这意味着当ph为10时脱羧最慢并因此是决定速度的部分反应。
出于有必要为了设定ph为9而必须使用ches或者其他比用于ph8的kpi缓冲剂明显毒性更大的水性缓冲剂,尽管转化率相同,但发明人仍视ph8为实施根据本发明方法的最佳ph值。
实施例5至8
与共溶剂在ph8时的一锅反应
因此,在ph为8时重复上述实施例2,其中以变化的量混入共溶剂且确定最终产物,对乙烯基苯酚4a的量。在实施例5中为了对比的目的而在没有共溶剂的情况下重复实施例2,而在实施例6中dmso作为可与水混溶的溶剂(5vol.-%)的代表,在实施例7中乙醚作为不可与水混溶的溶剂(5vol.-%)的代表,并在实施例8中,分别基于水性缓冲溶液的体积加入双倍量的乙醚(10vol.-%)。
图7示出了这些实验的结果。从中可见,可与水混溶的dmso的存在对产物4a的转化实际上没有造成不同,而不可与水混溶的乙醚在6小时反应时间后以10vol.-%的量使转化提高了超过10个百分点,但以5vol.-%的量甚至提高了20个百分点。
不希望受限于特定的理论,发明人认为转化率的提高可归因于两相的形成,其中四种不同苯酚的某些部分估计因为其较低的极性和亲水性(主要是反应物1a和最终产物4a的部分)而溶于醚相。由此推测在步骤b)和c)中单一反应间可能的抑制作用得以限制和/或总反应的化学平衡进一步向右移动。用双倍量的乙醚并未实现大幅提高转化率支持了抑制作用得以限制这一说法,这明显地归因于过量的苯酚、可能地过量的反应物溶于有机相中,使得反应速率降低。
实施例9
一锅反应的最佳条件
在成功优化ph值和共溶剂后,以如下条件重复实施例7中的实验:1a(23mm);丙酮酸盐(46mm);tpl(4mg);tal(20mg);fad(5mg);kpi缓冲剂(50mm),ph8;5vol.-%et2o,时间:6h;30℃;850转/分钟时摇动;并在此借助hplc以时间间隔确定反应物1a和产物2a至4a的量。
图8示出了该实验的结果,其中反应物1a的量在开始时非常快速地下降,并在6小时后所获得的向最终产物4a的转化率超过95%。
实施例10至20
制备不同的衍生物
应用或略微变化实施例9的最佳反应条件,改变了取代模式(有时也有起始底物,即苯酚1的量),如在随后的表1中所指示的,并且分别在24小时和48小时后借助hplc来确定转化率,即反应结束后的相对含量,并且同样在表格中指示。
表1
备注a:双倍量的tal
备注b:双倍反应时间(48小时)
可见,对于各个产物4的转化率不依赖于取代基r的类型(吸电子或推电子)和数目(一个或两个)始终在97%之上,并且三个部分反应的反应物分别以小于1%存在。
本领域技术人员将理解,反应将以类似完全的方式在起始苯酚1处以不同的取代模式进行,除非取代基明显地干扰酶促反应。
后处理:
通过加入饱和水性nh4cl溶液来停止反应,并以乙酸乙酯(3x15ml)萃取。在na2so4上干燥清洗后的有机相,并减压蒸发。借助快速色谱法(20%乙酸乙酯/己烷)纯化残留物,将包含各自乙烯基苯酚的级分合并并且在100mmhg下蒸发,这将获得与理论值的80-90%的转化率一致的作为无色油的所期望的对乙烯基苯酚。下面将给出如此制备的对乙烯基苯酚的光谱数据。
2-氟-4-乙烯基苯酚4a:
1h-nmr(300mhz,meod):δh[ppm]=5.10(d,3j2′c,1′=10.9hz,1h,2′-hc),5.59(dd,2j2′t,2′c=0.8hz,3j2′t,1′=17.6hz,1h,2′-ht),6.59(dd,3j1′,2′c=10.9hz,3j1′,2′t=17.6hz,1h,1′-h),6.85(dd,4j6,f=8.8hz,3j6,5=8.5hz,1h,6-h),7.02(dd,4j5,3=2.3hz,3j5,6=8.4hz,1h,5-h),7.14(dd,4j3,5=2.1hz,3j3,f=12.4hz,1h,3-h).
13c-nmr(75mhz,meod):δc[ppm]=112.3(c-2′),114.1(d,2j3,f=18.9hz,c-3),118.5(d,3j6,f=3.1hz,c-6),123.7(d,4j5,f=3.1hz,c-5),131.6(d,3j4,f=6.2hz,c-4),136.9(c-1′),145.9(d,2j1,f=13.3hz,c-1),152(d,1j2,f=240.1hz,c-2).19f(282mhz,meod)δf[ppm]=-139.7(dd,4jf,6=9.1hz,4jf,3=12.5hz).
2-氯-4-乙烯基苯酚4b:
1h-nmr(300mhz,meod):δh[ppm]=5.06(dd,2j2′c,2′t=0.7hz,3j2′c,1’=10.9hz,1h,2′-hc),5.56(dd,2j2′t,2′c=0.8hz,3j2′t,1′=17.6hz,1h,2′-ht),6.55(dd,3j1′,2′c=10.9hz,3j1′,2′1=17.6hz,1h,1′-h),6.81(d,3j6,5=8.4hz,1h,6-h),7.15(dd,4j5,3=2.2hz,3j5,6=8.4hz,1h,5-h),7.32(d,4j3,5=2.2hz,1h,3-h).
13c-nmr(75mhz,meod):δc[ppm]=112.4(c-2′),117.5(c-6),121.8(c-2),126.8(c-3),128.6(c-5),132.0(c-4),136.7(c-1′),154(c-1).
2-溴-4-乙烯基苯酚4c:
1h-nmr(300mhz,meod):δh[ppm]=5.07(dd,2j2′c,2′t=0.6hz,3j2′c,1′=11.0hz,1h,2′-hc),5.57(dd,2j2′t,2′c=0.6hz,3j2′t,1′=17.6hz,1h,2′-ht),6.55(dd,3j1′,2′c=10.9hz,3j1′,2′t=17.6hz,1h,1′-h),6.84(d,3j6,5=8.4hz,1h,6-h),7.21(dd,4j5,3=2.1hz,3j5,6=8.4hz,1h,5-h),7.51(d,4j3,5=2.1hz,1h,3-h).
13c-nmr(75mhz,meod):δc[ppm]=110.9(c-2),112.4(c-2′).117.1(c-6),127.4(c-5),131.7(c-3),132.3(c-4),136.4(c-1′),154.9(c-1).
2-甲基-4-乙烯基苯酚4d:
1h-nmr(300mhz,meod):δh[ppm]=2.17(s,3h,ch3),4.99(dd,2j2′c,2′t=1.1hz,3j2′c,1′=10.9hz,1h,2′-hc),5.53(dd,2j2′t,2′c=1.1hz,3j2′t,1′=17.7hz,1h,2′-ht),6.58(dd,3j1′,2′c=10.9hz,3j1′,2′t=17.6hz,1h,1′-h),6.68(d,3j6,5=8.2hz,1h,6-h),7.06(dd,4j5,3=2.2hz,3j5,6=8.2hz,1h,5-h),7.13(d,4j3,5=2.1hz,1h,3-h).
13c-nmr(75mhz,meod):δc[ppm]=16.2(ch3anc-2),110.4(c-2′),115.5(c-6),125.5(c-2),125.8(c-5),129.6(c-3),130.6(c-4),138.1(c-1′),156.5(c-1).
3-氟-4-乙烯基苯酚4e:
1h-nmr(300mhz,meod):δh[ppm]=5.16(dd,2j2′c,2′t=1.4hz,3j2′c,1′=11.3hz,1h,2′-hc),5.64(dd,2j2′t,2′c=1.3hz,3j2′t,1′=17.8hz,1h,2′-ht),6.47(dd,4j2,6=2.4hz,3j2,f=12.5hz,1h,2-h),6.57(dd,4j6,2=2.5hz,3j6,5=8.6hz,1h,6-h),6.74(dd,3j1′,2′c=11.3hz,3j1′,2′t=17.8hz,1h,1′-h),7.35(dd,3j5,6=8.7hz,4j5,f=8.8hz,1h,5-h).
13c-nmr(75mhz,meod):δc[ppm]=103.5(d,2j2,f=25.0hz,c-2),112.6(d,4j2′,f=2.7hz,c-2′),113.4(d,4j6,f=4.8hz,c-6),117.8(d,3j4,f=12.6hz,c-4),128.8(d,3j1′,f=5.8hz,c-1′),130.3(d,3j5,f=3.5hz,c-5),159.9(d,2j2,f=11.9hz,c-1),162.3(d,1j3,f=247.4hz,c-3);19f(282mhz,meod):δf[ppm]=-119.8(dd,4jf,5=8.8hz,3jf,2=12.6hz)
3-氯-4-乙烯基苯酚4f:
1h-nmr(300mhz,meod):δh[ppm]=5.18(dd,2j2′c,2′t=1.2hz,3j2′c,1′=11.0hz,1h,2′-hc),5.60(dd,2j2′t,2′c=1.2hz,3j2′t,1′=17.5hz,1h,2′-ht),6.71(dd,4j6,2=2.5hz,3j6,5=8.6hz,1h,6-h),6.79(d,3j2,6=2.5hz,1h,2-h),6.98(dd,3j1′,2′c=11.0hz,3j1′,2′t=17.6hz,1h,1′-h),7.47(d,3j5,6=8.6hz,1h,5-h).
13c-nmr(75mhz,meod):δc[ppm]=113.8(c-2′),115.7(c-6),116.8(c-2),128.1(c-4),128.3(c-5),133.7(c-1′),134.4(c-3),159.2(c-1).
2,3-二氟-4-乙烯基苯酚4g:
1h-nmr(300mhz,meod):δh[ppm]=5.25(dd,2j2’c,2′t=1.1hz,3j2′c,1’=11.4hz,1h,2′-hc),5.70(dd,2j2′t,2′c=1.1hz,3j2′t,1′=17.8hz,1h,2′-ht),6.68(ddd,5j6,3f=2.0hz,4j6,2f=8.0hz,3j6,5=8.5hz,1h,6-h),6.72(dd,3j1′,2′c=11.3hz,3j1′,2′t=17.8hz,1h,1′-h),7.11(ddd,5j5,2f=2.3hz,4j5,3f=8.3hz,3j5,6=8.5hz,1h,5-h).
13c-nmr(75mhz,meod):δc[ppm]=113.7(dd,3j6,2f=3.2hz,4j6,3f=3.2hz,c-6),115.1(d,4j2′,3f=5.1hz,c-2′),119.2(dd,3j4,2f=1.1hz,2j4,3f=9.7hz,c-4),122.2(dd,3j5,3f=4.5hz,4j5,2f=4.5hz,c-5),129.6(dd,4j1′,2f=3.1hz,3j1′,3f=3.3hz,c-1′),141.8(dd,2j2,3f=14.5hz,1j2,2f=241.2hz,c-2),147.5(dd,3j1,3f=2.8hz,2j1,2f=10.2hz,c-1),150.7(dd,2j3,2f=10.9hz,1j3,3f=248.4hz,c-3);19f(282mhz,meod):δf[ppm]=-146.1(ddd,5j3f,6=2.0hz,4j3f,5=8.1hz,3j3f,2f=18.4hz,1f,fanc-3),-165.1(ddd,5j2f,5=2.4h,4j2f,6=8.0hz,3j2f,3f=18.3hz,1f,fanc-2).
3-氯-2-氟-4-乙烯基苯酚4h:
1h-nmr(300mhz,meod):δh[ppm]=5.27(d,3j2′c,1′=11.1hz,1h,2′-hc),5.68(dd,2j2′t,2′c=0.9hz,
3j2′t,1′=17.5hz,1h,2′-ht),6.85(dd,4j6,f=8.6hz,3j6,5=8.6hz,1h,6-h),6.96(dd,3j1′,2′c=11.0hz,3j1′,2′t=17.5hz,1h,1′-h),7.29(dd,5j5,f=2.0hz,3j5,6=8.7hz,1h,5-h).
13c-nmr(75mhz,meod):δc[ppm]=115.4(c-2′),117.1(d,3j6,f=3.1hz,c-6),121.5(d,2j3,f=15.1hz,c-3),122.2(d,4j5,f=4.0hz,c-5),129.2(d,3j4,f=1.3hz,c-4),132.9(d,4j1′,f=3.1hz,c-1′),146.8(d,2j1,f=13.3hz,c-1),148.9(d,1j2,f=241.2hz,c-2);19f(282mhz,meod):δf[ppm]=-139.6(dd,4jf,6=8.8hz,5jf,5=2.0hz).
为此,本发明提供了一种用于制备对乙烯基苯酚的一锅反应,根据该反应能够高效并经济地制备多种化合物,这在现有技术中甚至几乎不可能实现。
- 还没有人留言评论。精彩留言会获得点赞!