一种含碟烯基磷光铱配合物及其制备方法和电致发光器件与流程
本发明属于有机光电材料领域,尤其涉及一种含碟烯基磷光铱配合物及其制备方法和电致发光器件。
背景技术:
有机电致发光器件(organiclight-emittingdiodes,oleds)与液晶显示相比,具有驱动电压低、发光亮度和效率高、发光视角宽、响应速度快、器件厚度小、可柔性显示等众多优点,在信息显示和固态照明等领域有着巨大的应用前景,被誉为下一代“明星”平板显示技术。
根据自旋统计规律,有机电致发光材料中电荧光器件的最高内量子效率的上限为25%,这造成了有机电荧光器件的发光效率的徘徊不前,最大外量子效率只能达到5%左右。而近年来在有机电致发光材料研究上最具突破性的发展之一是对电激发磷光现象的发现,这些磷光材料主要包括一些重金属配合物,如锇、铂、铱的配合物,由于存在强烈的自旋轨道偶合,使得其配合物的单重态激子和三重态激子混杂。进而使得电致磷光可以不受自旋统计规律的影响,最大量子效率可达100%。因此,对这种可明显提高器件外量子效率的重金属配合物的研究具有重要基础理论意义的和巨大的实用价值。环金属铱配合物由于其相对较短的激发态寿命、高的发光量子效率、优异的发光颜色可调性以及易于制备的特点,被广泛应用于有机电致发光领域。
通常磷光材料自身的聚集会引起发光减弱,也就是通常所说的浓度淬灭效应。为了防止这些问题,铱配合物应用于器件时一般以低浓度掺杂到主体之中,目前绝大多数高效磷光oleds都是在较低的掺杂浓度(﹤10%)下实现的。但是这就使得器件的制备很繁琐且重复性差,器件的光色稳定性和长效性能都会受到影响,增加了产业化生产的难度和成本。因此设计合成在很宽的掺杂浓度范围内,甚至在非掺杂的器件结构中都能实现高性能(低驱动电压、高亮度、高效率)磷光发射的新型材料体系,无论对促进实验室基础研究还是对提高未来产业化的生产效率都有着深远的意义。通过超支化或在铱配合物配体上增加阻隔基团可以降低磷光淬灭,进而达到以上目的。其中超支化铱配合物的合成比较复杂,而且过多超支化也会因为空穴和电子不能在铱核上复合而使器件效率较低。而在铱配合物配体上增加紧凑的阻隔基团则是一种更为简便有效的方法,其中引入刚性、非共轭、同时具有大空间位阻的官能团的设计最为有效。现有技术中在铱配合物上引入了立体位阻的派烯结构,刚性的咔唑作为铱配合物的阻隔基团,用二苯胺作为铱配合物的阻隔基团,立体位阻芳基硅烷作为树枝合成铱配合物或在配体上紧密地接上一个具有很大立体位阻的多芳基,掺杂器件的最大流明效率为49.8cda-1。总体而言,由于大部分位阻的刚性不够或者间隔的空间不够,能真正用于高效非掺杂有机电致发光器件的还不多。
三蝶烯(triptycene)是由三个苯环通过桥头碳原子之间相互铰链而成的五环化合物,具有独特的结构特点(一般的阻隔基团是单键连接,而三蝶烯是双键连接)和丰富的反应性能,在分子机器、材料化学、超分子化学以及有机催化等很多领域引起了人们越来越多的关注。特别是这种独特的三位刚性结构中,三个芳环是通过饱和碳原子相连,彼此没有形成共轭,位阻面更大,能够抵御外界的接触,三个芳环能形成巨大的立障空间。如果能将该结构融于铱配合物的设计合成中将会带来诸多优点,非常契合高效率铱配合物的特点。目前文献中也有关于碟烯基铱配合物的报道(acscatal.2014,4,3411-3420,inorg.chem.2013,52,8653-8664),但是,这些配合物要么没有发光性能,要么就是离子型的,不适合于在有机电致发光中应用。
《三蝶烯羧酸-过渡金属配合物的合成与物化性质研究》作者:谭爱东,华侨大学。其不足之处在于,该配合物不可能在oled器件上应用,三蝶烯在该配合物中仅起到骨架的作用。
技术实现要素:
1.发明要解决的技术问题
针对现有技术的碟烯基铱配合物不适合于在有机电致发光器件中应用的问题,本发明提供了一种含碟烯基磷光铱配合物及其制备方法和电致发光器件。本发明以含碟烯结构的配体与铱配位,获得碟烯包裹的磷光材料,并将其作为掺杂于发光层,制成的oled器件可实现掺杂浓度高,磷光淬灭小,光谱稳定性好,效率高的效果。
2.技术方案
为解决上述问题,本发明提供的技术方案为:
一种含碟烯基的磷光铱配合物,该配合物结构式通式(1)所示:
其中,y为硫、氧或取代氮,l^x为含氮辅助配体。
优选地,所述配合物的具体结构为:
其中,r为取代氮,取代基为氢,1到8个原子的烷基或苯基。
一种如以上所述的含碟烯基铱配合物的制备方法:
a、将1-醛基三碟烯溶于干燥的有机溶剂中,在惰性气体保护、催化剂存在的条件下与邻氨基化合物进行缩合反应,得到含碟烯结构的环金属化配体;
b、将ircl3溶于水中,加入含碟烯结构的环金属化配体和有机溶剂,惰性气体保护搅拌,得铱的二氯桥中间体;
c、将铱的二氯桥中间体溶于有机溶剂中,与辅助配体(l^x)在碱的作用下,反应温度控制在20~200℃,惰性气体保护下搅拌1~24h,得含碟烯基铱配合物。
优选地,所述的步骤a的反应物用量是,按摩尔分数计,1-醛基三叠烯1份,邻氨基化合物1~4份,有机溶剂50~500份,催化剂0.1~10份。
优选地,所述的步骤b的反应物用量是,按摩尔分数计,ircl31份,含碟烯结构的环金属化配体2~4份,有机溶剂50~500份,步骤b中所述的有机溶剂是甲氧基乙醇、乙氧基乙醇、乙二醇、丙二醇、甘油或缩水甘油醚。
优选地,所述的步骤c的反应物用量是,按摩尔分数计,铱的二氯桥化合物1份,辅助配体1~5份,碱1~5份,有机溶剂50~500份,步骤c中所述的有机溶剂是二氯甲烷、甲氧基乙醇、乙氧基乙醇、乙二醇、丙二醇、甘油或缩水甘油醚;所述的碱为碳酸钾、碳酸氢钾、碳酸钠、碳酸氢钠、乙酸钠、三乙胺或吡啶。
优选地,所述的步骤a式中y是硫、氧或取代氮,取代基为氢,1~8个碳的烷基或苯基;所述的有机溶剂是乙酸、n,n-二甲基甲酰胺,二甲基亚砜,乙醇,乙氧基乙醇;所述的催化剂为无水硫酸镁或三氟乙酸。
优选地,步骤a中在20~150℃下进行缩合反应,反应2h~24h,得到含碟烯结构的环金属化配体。
优选地,步骤b的反应温度控制在50~200℃,惰性气体保护搅拌8~24h,得铱的二氯桥中间体,温度和时间限定是为了提高产率。
一种电致发光器件,发光层含有以上所述的含碟烯基铱配合物磷光材料。
3.有益效果
采用本发明提供的技术方案,与现有技术相比,具有如下有益效果:
(1)本发明碟烯基的引入使铱配合物的浓度淬灭明显减少,因此器件掺杂浓度大大提高,即使非掺杂时也有较高的效率;
(2)本发明在浓度或电压变化时器件的光谱变化减少,oled器件光色稳定性好;
(3)本发明磷光材料制成的有机电子发光器件驱动电压低至2v,亮度高达26370cd/m2,最大电流效率和外量子效率分别为30cd/a和11.7%;
(4)本发明的发光材料发光量子效率在60%以上,在低掺杂浓度时高于没有碟烯结构的铱配合物,且在高浓度时优势更加明显;
(5)本发明的结构原理简单、制作成本低、易于实现。
附图说明
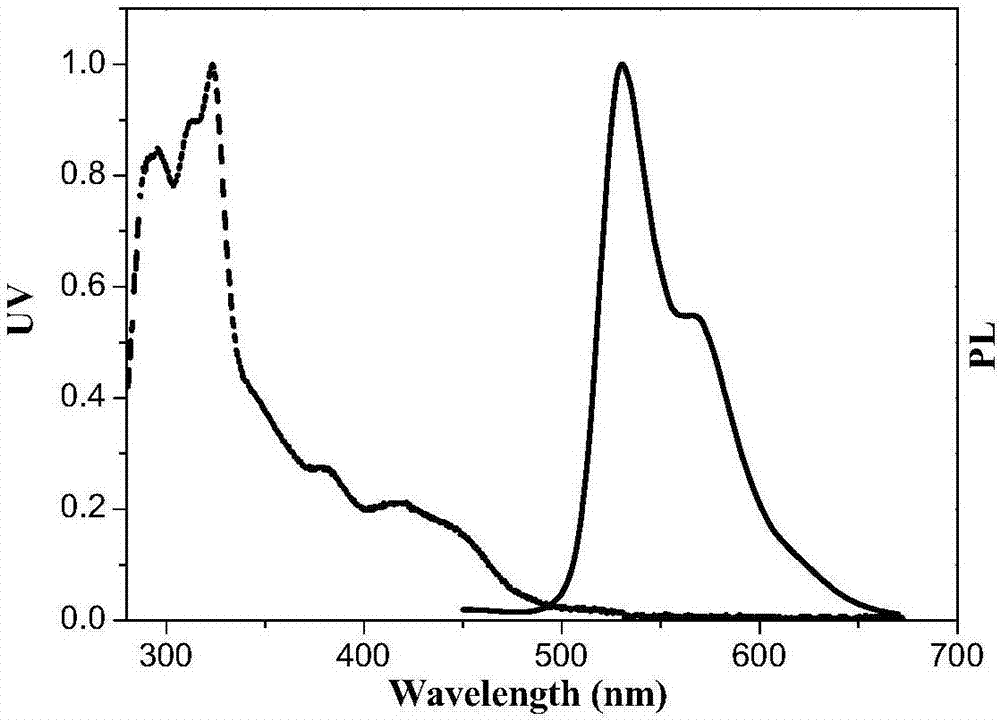
图1磷光铱配合物((tps)2ir(tp))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图2磷光铱配合物((tps)2ir(tp))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图3磷光铱配合物((tps)2ir(pic))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图4磷光铱配合物((tps)2ir(bpz))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图5磷光铱配合物((tps)2ir(qpz))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图6磷光铱配合物((tpi)2ir(pic))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图7磷光铱配合物((tpi)2ir(tp))在二氯甲烷溶液中的紫外可见吸收(uv/vis)光谱,光致发光(pl)光谱;
图8磷光铱配合物电致发光器件的结构示意图;
图9实施例13中电致发光器件的亮度-电流效率图;
图10实施例13中电致发光器件的亮度-外量子效率图;
图11为本发明的含碟烯基的磷光铱配合物的结构通式。
具体实施方式
以下实施例是对本发明的进一步说明,不是对本发明的限制。
图1-7中,虚线表示吸收峰,实线表示发射峰。
一种含碟烯基的磷光铱配合物,该配合物结构式通式(1)所示:
其中,y为硫、氧或取代氮,l^x为含氮辅助配体。
当y为取代氮时,所述配合物的具体结构为:
其中,r为取代氮,取代基为氢,1到8个原子的烷基或苯基,如甲基、乙基、异丙基、叔丁基、苯基、2,6-二甲基苯基、2,4,6-三甲基苯基。
一种如以上所述的含碟烯基铱配合物的制备方法:
a、将1-醛基三碟烯溶于干燥的有机溶剂中,在惰性气体保护(本发明中的惰性气体均可选择氮气、氦气和氩气,常选择使用氮气)、催化剂存在的条件下与邻氨基化合物进行缩合反应,得到含碟烯结构的环金属化配体;
以上反应物用量是,按摩尔分数计,1-醛基三叠烯1份,邻氨基化合物1~4份,有机溶剂50~500份,催化剂0.1~10份。在具体应用时,1-醛基三叠烯、邻氨基化合物、有机溶剂和催化剂三者之间的摩尔分数之比可以为1:1:50:0.1、1:2:60:0.2、1:2.5:100:1、1:4:200:1.5、1:3:500:10或1:3.5:400:8等数值。以上反应式中y是硫、氧或取代氮,取代基为氢,1~8个碳的烷基或苯基;所述的有机溶剂是乙酸,n,n-二甲基甲酰胺,二甲基亚砜,乙醇或乙氧基乙醇中的一种或几种组合;所述的催化剂为无水硫酸镁或三氟乙酸的一种或两种组合。在20~150℃(具体应用时,可为20℃、30℃、35℃、50℃、70℃、80℃、100℃、130℃或150℃等数值)下进行缩合反应,反应2h~24h(具体应用时,可为2h、3h、5h、10h、15h、18h、20h、24h或23h等数值),得到含碟烯结构的环金属化配体。
b、将ircl3溶于水中,加入含碟烯结构的环金属化配体和有机溶剂,惰性气体保护搅拌,得铱的二氯桥中间体;
反应物用量是,按摩尔分数计,ircl31份,含碟烯结构的环金属化配体2~4份(具体应用时,可以为2、2.5、3、3.6、3.9或4份等数值),有机溶剂50~500份(具体应用时,可以为50、60、70、80、88、90、100、120、200、300、00、500或450份等数值),步骤b中所述的有机溶剂是甲氧基乙醇、乙氧基乙醇、乙二醇、丙二醇、甘油或缩水甘油醚。反应温度控制在50~200℃(具体应用时,可为50℃、180℃、160℃、135℃、70℃、80℃、100℃、190℃或200℃等数值),惰性气体保护搅拌8~24h(具体应用时,可为8h、9h、13h、15h、10h、15h、18h、20h、24h或23h等数值),得铱的二氯桥中间体,温度和时间限定是为了提高产率。
c、将铱的二氯桥中间体溶于有机溶剂中,与辅助配体(l^x)在碱的作用下,反应温度控制在20~200℃(具体应用时,可为20℃、30℃、35℃、50℃、70℃、80℃、100℃、130℃、180℃、160℃、190℃或200℃等数值),惰性气体保护下搅拌1~24h(具体应用时,可为1h、2h、3h、5h、10h、15h、18h、20h、24h或23h等数值),得含碟烯基铱配合物。
反应物用量是,按摩尔分数计,铱的二氯桥化合物1份,辅助配体1~5份(具体应用时,可以为1、2、2.5、3、3.6、5、3.9或4份等数值),碱1~5份(具体应用时,可以为1、2、2.5、3、3.6、5、3.9或4份等数值),有机溶剂50~500份(具体应用时,可以为50、60、70、80、88、90、100、120、200、300、00、500或450份等数值),步骤c中所述的有机溶剂是二氯甲烷、甲氧基乙醇、乙氧基乙醇、乙二醇、丙二醇、甘油或缩水甘油醚种的一种或几种组合;所述的碱为碳酸钾、碳酸氢钾、碳酸钠、碳酸氢钠、乙酸钠、三乙胺或吡啶的一种或几种组合。
一种电致发光器件,发光层105中含有以上所述的含碟烯基铱配合物磷光材料。
实施例1
本实施例的一种的含碟烯基铱配合物的制备方法,在120ml封管中加入2.00g(7.09mmol)1-醛基三蝶烯,0.87g(7.09mmol)邻氨基苯硫酚及15ml(194mmol)无水n,n-二甲基甲酰胺。加热至120℃,在氮气环境下反应24h。反应完毕,加20ml水后,乙酸乙酯萃取,有机相水洗后用无水硫酸钠干燥并旋干,用石油醚:二氯甲烷=1:1过柱,旋干后得到2-(1-三碟烯基)苯并噻唑环金属化配体的白色固体2.30g,产率80%。1hnmr(400hz,cdcl3)δ5.64(s,1h),7.06(s,1h),7.10-7.16(m,5h),7.51-7.63(m,7h),7.68(t,j=8hz,1h),8.05(d,j=8hz,1h),8.39(d,j=8hz,1h)。
取0.77g(2mmol)2-(1-三叠烯基)苯并噻唑,0.35g(1mmol)三水合三氯化铱加入到封管中,然后向封管加入30ml(310mmol)乙二醇乙醚及10ml(556mmol)水。加热至120℃并在n2环境下反应24h。反应完后冷却至室温,过滤,并用适量乙醇冲洗滤饼,将滤饼干燥得到橙黄色铱二氯桥中间体固体。
将0.20g(0.1mmol)二氯桥中间体、0.043g(0.2mmol)3-三氟甲基-5-(2-吡啶基)-1,2,4-三唑和0.14g(1mmol)碳酸钾加入到封管中,然后向封管中加入20ml(312mmol)二氯甲烷,在60℃下反应5h。反应完毕,用柱色谱分离产物,得到(tps)2ir(tp)橙红色固体0.025g,产率10.2%。(tps)2ir(tp)的核磁图谱为:1hnmr(400hz,cdcl3)δ5.22(s,1h),5.28(s,1h),5.30(s,2h),5.90(d,j=7.8hz,1h),5.98(d,j=7.5hz,1h),6.13(d,j=8.6hz,1h),6.43(s,2h),6.70-6.82(m,3h),6.85-7.07(m,9h),7.14(t,j=8.0hz,1h),7.22(d,j=6.6hz,1h),7.27-7.53(m,10h),7.73(t,j=7.2hz,1h),7.85-7.95(m,2h),8.10(d,j=7.2hz,1h)。19fnmr(cdcl3,367mhz)δ-63.19(s,3f)。
为说明碟烯基的特点制备了参比物:0.42g(2mmol)2-苯基苯并噻唑和0.35g(1mmol)三水合三氯化铱放入100ml封管中,然后向封管加入30ml乙二醇乙醚及10ml水。加热至120℃并在n2环境下反应24h。反应完后冷却至室温,过滤,并用适量乙醇冲洗滤饼,将滤饼干燥得到橙黄色铱二氯桥中间体固体。再将所得二氯桥、0.43g(2mmol)3-三氟甲基-5-(2-吡啶基)-1,2,4-三唑和1.4g(10mmol)碳酸钾加入到封管中,然后向封管中加入60ml二氯甲烷,在60℃下反应5h。反应毕,用柱色谱分离产物。得到(ps)2ir(tp)橙红色固体。(ps)2ir(tp)的核磁图谱为:1hnmr(400hz,cdcl3)δ6.15(d,j=4.9hz,1h),6.39(d,j=4.9hz,1h),6.50(d,j=6.7hz,1h),6.66(d,j=8.2hz,1h),6.75(t,j=4.1hz,1h),6.82(t,j=7.4hz,1h),6.91-7.06(m,3h),7.13-7.24(m,2h),7.27-7.33(m,4h),7.69-7.88(m,5h),8.24(d,j=4.1hz,1h)。19fnmr(cdcl3,367mhz)δ-63.17(s,3f)。
实施例2
本实施例的一种的含碟烯基铱配合物的制备方法,将0.20g(0.1mmol)二氯桥中间体、0.05g(0.4mmol)2-吡啶甲酸和0.14g(1mmol)碳酸钾加入到封管中,向封管中加入20ml二氯甲烷,加热至60℃反应5h。反应毕,用柱层析分离产物,得到(tps)2ir(pic)橙红色固体0.023g,产率16%(产率跟反应原料有关,不同原料产率不同。)。(tps)2ir(pic)的核磁图谱为:1hnmr(400hz,cdcl3)δ5.17(s,1h),5.27(s,1h),5.73(d,j=9.7hz,1h),6.08-6.16(m,2h),6.41(d,j=7.8hz,2h),6.66(d,j=7.9hz,1h),6.72(d,j=8.0hz,1h),6.86-7.10(m,9h),7.17-7.25(m,2h),7.30-7.55(m,10h),7.66-7.80(m,2h),7.90-8.00(m,2h),8.10(d,j=7.4hz,1h),8.40-8.46(m,1h)。
实施例3
本实施例的一种的含碟烯基铱配合物的制备方法,将0.20g(0.1mmol)二氯桥中间体、0.054g(0.2mmol)3-三氟甲基-5-(2-(4-叔丁基)吡啶基)吡唑和0.14g(1mmol)碳酸钾加入到封管中,然后向封管中加入20ml二氯甲烷,在60℃下反应5h。反应毕,用柱色谱分离产物。得到(tps)2ir(bpz)橙红色固体0.019g,产率15%.(tps)2ir(bpz)的核磁图谱为:1hnmr(400hz,cdcl3)δ1.25(s,9h),5.22(s,1h),5.26(s,1h),5.92(d,j=7.6hz,1h),5.98(d,j=7.5hz,1h),6.07(d,j=8.5hz,1h),6.45(d,j=4.8hz,2h),6.70-6.84(m,6h),6.87-7.06(m,9h),7.16(t,j=7.3hz,1h),7.20(s,1h),7.28-7.39(m,5h),7.42-7.50(m,4h),7.51(s,1h),7.88(t,j=8.2hz,2h)。19fnmr(cdcl3,367mhz)δ-59.81(s,3f)。
实施例4
本实施例的一种的含碟烯基铱配合物的制备方法,将0.20g(0.1mmol)二氯桥中间体、0.085g(0.2mmol)3-三氟甲基-5-(1-(4-(2,6-二异丙基苯基))异喹啉基)吡唑和0.14g(1mmol)碳酸钾加入到封管中,然后向封管中加入20ml二氯甲烷,在60℃下反应5h。反应毕,用柱色谱分离产物。得到(tps)2ir(qpz)橙红色固体0.017g,产率12%。(tps)2ir(qpz)的核磁图谱为:1hnmr(400hz,cdcl3)δ1.20-1.30(m,12h),2.15-2.33(m,2h),5.10(s,1h),5.23(s,1h),5.93(d,j=7.6hz,1h),6.04(d,j=7.5hz,1h),6.27(d,j=7.8hz,2h),6.43(s,1h),6.63(dd,j=8.2hz,2h),6.75(dd,j=7.4hz,2h),6.82-6.96(m,6h),6.88-7.00(m,3h),7.08(d,j=7.6hz,1h),7.13-7.25(m,9h),7.27-7.35(m,3h),7.43(t,j=7.2hz,2h),7.47-7.57(m,3h),7.61(t,j=7.4hz,1h),7.80-7.89(m,2h),8.70(d,j=8.8hz,1h)。19fnmr(cdcl3,367mhz)δ-59.98(s,3f)。
实施例5
本实施例的一种的含碟烯基铱配合物的制备方法,在120ml封管中加入2.00g(7.09mmol)1-醛基三蝶烯,1.30g(7.09mmol)邻氨基二苯胺,1.20g(10mmol)无水硫酸镁及30ml冰醋酸。加热至120℃,在氮气环境下反应12h。反应完毕,加50ml水后,乙酸乙酯萃取,有机相水洗后用无水硫酸钠干燥,并旋干,用柱色谱分离产物,旋干后得到1-苯基-2-(1-三碟烯基)苯并咪唑的白色固体1.01g,产率32.4%。1hnmr(400hz,cdcl3)δ5.51(s,1h),6.16(s,1h),6.75-6.88(m,2h),7.01-7.08(m,4h),7.15-7.22(m,2h),7.25-7.32(m,3h),7.40-7.51(m,8h),8.10(d,j=8hz,1h)。
取0.89g(2mmol)1-苯基-2-(1-三碟烯基)苯并咪唑,0.35g(1mmol)三水合三氯化铱加入到封管中,然后向封管加入30ml乙二醇乙醚及10ml水。加热至120℃并在n2环境下反应24h。反应完后冷却至室温,过滤,并用适量乙醇冲洗滤饼,将滤饼干燥得到橙黄色铱二氯桥中间体固体。
将0.55g(0.25mmol)二氯桥b、0.10g(0.8mmol)2-吡啶甲酸和0.14g(1mmol)碳酸钾加入到封管中,向封管中加入30ml二氯甲烷,加热至60℃反应10h。反应毕,用柱层析分离产物,得到(tpi)2ir(pic)橙红色固体0.18g,产率35%。(tpi)2ir(pic)的核磁图谱为:1hnmr(400hz,cdcl3)δ5.03(s,1h),5.14(s,1h),5.54(d,j=5.2hz,2h),6.03-6.10(m,3h),6.62-6.70(m,5h),6.74-6.80(m,10h),6.88-6.93(m,2h),6.96-7.01(m,4h),7.11(d,j=7.1hz,2h),7.17-7.24(m,4h),7.31-7.34(m,3h),7.37-7.40(m,3h),7.67-7.77(m,4h),8.10-8.13(m,1h),8.21(d,j=8.5hz,1h)。
实施例6
本实施例的一种的含碟烯基铱配合物的制备方法,将0.55g(0.25mmol)二氯桥b、0.11g(0.5mmol)3-三氟甲基-5-(2-吡啶基)-1,2,4-三唑和0.14g(1mmol)碳酸钾加入到封管中,向封管中加入30ml二氯甲烷,加热至60℃反应10h。反应毕,用柱层析分离产物,得到(tpi)2ir(tp)橙红色固体0.16g,产率30.5%。(tpi)2ir(tp)的核磁图谱为:1hnmr(400hz,cdcl3)δ5.05(s,1h),5.24(s,1h),5.59(s,1h),5.66-5.75(m,4h),5.87(d,j=7.9hz,1h),6.43-6.47(m,2h),6.59(d,j=5.4hz,1h),6.65-6.78(m,7h),6.80-6.92(m,5h),6.95-7.01(m,3h),7.03-7.17(m,7h),7.22(d,j=8.3hz,2h),7.30-7.33(m,2h),7.36-7.44(m,3h),7.60-7.73(m,4h),8.0-8.05(m,2h)。19fnmr(cdcl3,367mhz)δ-63.01(s,3f)。
实施例7
本实施例的一种的含碟烯基铱配合物的制备方法,配合物(tps)2ir(tp)及对比配合物(tps)2ir(tp)的光物理特性研究。图1所示是配合物(tps)2ir(tp)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=332nm,发射峰位λmax=542nm。该配合物以1%和30%的质量比掺杂于聚甲基丙烯酸甲酯后测得的光致发光量子效率为分别为68.3%和46.3%。图2所示是配合物(ps)2ir(tp)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=323nm,发射峰位λmax=531nm。该配合物以1%和30%的质量比掺杂于聚甲基丙烯酸甲酯后测得的光致发光量子效率为分别为60.1%和14.4%。由结果可知,有三碟烯结构的铱配合物发光波长红移11nm,红移较少,为烷基取代导致的正常现象。1%掺杂浓度时发光量子效率有三碟烯结构的铱配合物高8.2%,30%掺杂浓度时发光量子效率高31.9%,表明有三碟烯结构的铱配合物发光量子效率较高,特别是在高浓度时优势更加明显,体现了三碟烯铱配合物在抑制浓度淬灭时的优点。
实施例8
本实施例是对含碟烯基铱配合物(tps)2ir(pic)的光物理特性研究,图3所示是配合物(tps)2ir(pic)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=332nm,发射峰位λmax=554nm。
实施例9
本实施例是对含碟烯基铱配合物(tps)2ir(bpz)的光物理特性研究。图4所示是配合物(tps)2ir(bpz)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=332nm,发射峰位λmax=546nm。
实施例10
本实施例是对含碟烯基铱配合物(tps)2ir(qpz)的光物理特性研究。图5所示是配合物(tps)2ir(qpz)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=332nm,发射峰位λmax=551nm。
实施例11
本实施例是对含碟烯基铱配合物(tpi)2ir(pic)的光物理特性研究。图6所示是配合物(tpi)2ir(pic)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=306nm,发射峰位λmax=538nm。
实施例12
本实施例是对含碟烯基铱配合物(tpi)2ir(tp)的光物理特性研究。图7所示是配合物(tpi)2ir(tp)在二氯甲烷中形成的稀溶液的吸收和发射光谱。该配合物的吸收峰位λmax=302nm,发射峰位λmax=526nm。
实施例13
本实施例是含碟烯基铱含配合物(tps)2ir(tp)的有机电致发光器件的制作方法。将ito玻璃相机地在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(120℃),再将ito玻璃做30秒的氧等离子体处理,传送到真空室内制备有机膜和金属电极。本实验的器件结构为:ito/moo3(1nm)/mcp(40nm)/mcp:(tps)2ir(tp)(8wt%,20nm)/bphen(40nm)/lif(1nm)/al。mcp为9,9′-(1,3-苯基)二-9h-咔唑,bphen为4,7-二苯基-1,10-邻二氮杂菲。器件的开启电压为2v;器件的最大电流效率(图9)和外量子效率(图10)分别为30cd/a和11.7%。
实施例14
本实施例中的一种电致发光器件,图8公开了本发明所述电致发光器件100的结构示意图,其中:含有多层薄膜结构,包括由下至上依次设置的基片层101、阳极102、空穴注入层或者空穴传输层103、第一缓冲层或者第一阻挡层104、发光层105、第二缓冲层或者第二阻挡层106、电子注入层或者电子传输层107、阴极108;另外,该有机电致发光器件100中,发光层105含有实施例1-12中所述的磷光铱配合物。
以上示意性的对本发明及其实施方式进行了描述,该描述没有限制性,附图中所示的也只是本发明的实施方式之一,实际的结构并不局限于此。所以,如果本领域的普通技术人员受其启示,在不脱离本发明创造宗旨的情况下,不经创造性的设计出与该技术方案相似的结构方式及实施例,均应属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!