一种噁二唑次膦酰亚胺铱配合物及制备方法与应用与流程
本发明涉及一种有机电致发光材料,及其在有机电致发光器件中的应用,属于有机电致发光显示技术领域。
背景技术:
有机发光二极管(Organic Light-Emitting Diodes,以下简称OLED)由于具有超轻薄、响应速度快、自发光、全固化、温度特性好、可实现柔软显示等特性,在各种领域有着广泛的应用。
1963年Pope等人(J.Chem.Phys.1963,38:2042-2043)研究了基于蒽单晶片(10~20μm)的蓝色电致发光器件,因蒽单晶发光层较厚和所使用的电极材料(银胶和氯化钠溶液)的制约,器件的发光启动电压高达400V,且效率和亮度均较低。然而,该发现开辟了发光科技的一个新领域。此后的二十多年间,OLED的研究进展缓慢。直至1987年,美国柯达公司的C.W.Tang等(Appl.Phys.Lett.1987,51:913-915)才取得了具有里程碑意义的突破。他们采用双层结构以8-羟基喹啉铝(Alq3)作发光层、芳香二胺作空穴传输层、ITO作阳极、Mg:Ag(10:1)合金作阴极的双层器件,得到较高量子效率(1%)和发光效率(1.5lm/W):高亮度(>1000cd/m2)和较低驱动电压(≤10V的器件。这一进展重新唤起了OLED应用于全色平板显示器的希望,材料和器件的研究迅速成为研究的热点。1988年,Adchi等人(J.Appl.Phys.1988,27:L269-L271)推出了多层夹心式结构,大大扩展了OLED材料的选择范围。
发光染料被认为是OLED器件的核心部件。根据发光染料的发光机理不同,可将其分为单线态(S1)发光的荧光材料和三线态(T1)发光的磷光材料。自旋统计规律表明,基于磷光染料的OLED的内量子效率可以达到100%,因而磷光染料已成为电致发光材料领域的研究热点[(a)Appl.Phys.Lett.2000,77:904-906.(b)J.Am.Chem.Soc.2001,123:4304-4312.(c)Nature 2003,421:829-833.(d)Adv.Mater.2005,17:1109-1121.(e)Coord.Chem.Rev.2009,253:1709-1758.(f)Adv.Mater.2011,23:926-952.(g)Angew.Chem.,Int.Ed.2012,51:8178-8211.(h)J.Am.Chem.Soc.2013,135:14321-14328]。然而,对于磷光OLED器件而言,往往存在着因三线态-三线态湮灭(TTA)和三线态-极化子淬灭(TPQ)所导致的效率滚降现象,即随着发光亮度和电流密度的增加器件效率急剧下降。其主要原因在于:一方面,对于磷光染料来说,其发光寿命较长(微秒~毫秒级),激发态不能快速回到基态,导致发光淬灭,使器件的发光效率下降;另一方面,配合物发光染料的载流子传输性能较差,不能有效地传输电子传输层和空穴传输层输运过来的电子和空穴载流子,使得载流子和激子聚集,以致浓度过大使得器件效率急剧下降。这些问题严重阻碍了OLED在全色平板显示和固态白光照明领域的应用。
技术实现要素:
本发明的目的是提供一种噁二唑次膦酰亚胺铱配合物,以及采用该化合物材料为发光中心的有机电致发光器件及其制备方法。
本发明的技术方案如下:
一种噁二唑次膦酰亚胺铱配合物,所述噁二唑次膦酰亚胺铱配合物噁二唑次膦酰亚胺铱配合物为二[7,8-苯并喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱,结构式如1所示;二[2-苯基喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱,结构式如2所示;二[1-苯基异喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱,结构式如3所示。
所述的噁二唑次膦酰亚胺铱配合物的制备方法,其特征在于:在无水无氧条件下,将双-[7,8-苯并喹啉]合二氯化铱和2.5倍当量的2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑的钾盐溶于乙二醇单乙醚中,于120-150℃反应10-15小时,过滤,重结晶,最后通过柱层层析提纯得到配合物二[7,8-苯并喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱。
所述的噁二唑次膦酰亚胺铱配合物的制备方法,其特征在于:在无水无氧条件下,将双-[2-苯基喹啉]合二氯化铱和2.5倍当量的2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑的钾盐溶于乙二醇单乙醚中,于120-150℃反应10-15小时,过滤,重结晶,最后通过柱层层析提纯得到配合物二[2-苯基喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱。
所述的噁二唑次膦酰亚胺铱配合物的制备方法,其特征在于:在无水无氧条件下,将双-[1-苯基异喹啉]合二氯化铱和2.5倍当量的2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑的钾盐溶于乙二醇单乙醚中,于120-150℃反应10-15小时,过滤,重结晶,最后通过柱层层析提纯得到配合物二[1-苯基异喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱。
所述的噁二唑次膦酰亚胺铱配合物的制备方法,其特征在于:所述柱层层析柱的洗脱剂为石油醚和乙酸乙酯的混合物,石油醚和乙酸乙酯的体积比为5:1。
噁二唑次膦酰亚胺铱配合物在制备有机电致发光器件中的应用。
用1H NMR、质谱、元素分析(C、H、N)表征证实了这些化合物的结构,并测定了化合物的紫外吸收和发射光谱,检测所用仪器为Bruker DPX 400核磁共振仪,Esquire-LC_00136质谱仪,Exeter Analytical CE-440型元素分析仪,Agilent Cary 60紫外-可见分光光度计以及Hitachi F-7000荧光光谱仪。
以配合物1、2和3为发光中心的器件结构,其结构和所用材料的结构如下:
采用ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物1、2或3(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)电致发光器件结构。其中HIO2作为空穴注入层,NPB为空穴传输层,TCTA 为激子阻挡层,mCP作为发光配合物染料的主体材料,TPBI为电子传输和空穴阻挡层,Liq为电子注入层。基于配合物1的电致发光器件(D1)的最大发射峰位于534nm,其色坐标为(CIE,x=0.36,y=0.60),为黄光,器件的最大发光亮度、电流效率和外量子效率分别为29800cd/m2、60.77cd/A和18.3%。基于配合物2的电致发光器件(D2)的最大发射峰位于588nm,其色坐标为(CIE,x=0.57,y=0.42),为橙光,器件的最大发光亮度、电流效率和外量子效率分别为19340cd/m2、25.99cd/A和13.7%。基于配合物3的电致发光器件(D3)的最大发射峰位于613nm,其色坐标为(CIE,x=0.64,y=0.33),为红光,器件的最大发光亮度、电流效率和外量子效率分别为7007cd/m2、7.56cd/A和8.17%。随着发光亮度的增大器件的发光效率下降不明显,表现为较弱的效率滚降。器件的上述优良性能表明这三种配合物染料在有机电致发光器件中具有应用价值。
本发明的有益效果是:本发明配体中含有两种电子传输基团(即噁二唑和膦氧基),可提高此种配合物染料的载流子传输性能,从而提高其电致发光器件的发光效率,改善其效率滚降现象。
附图说明
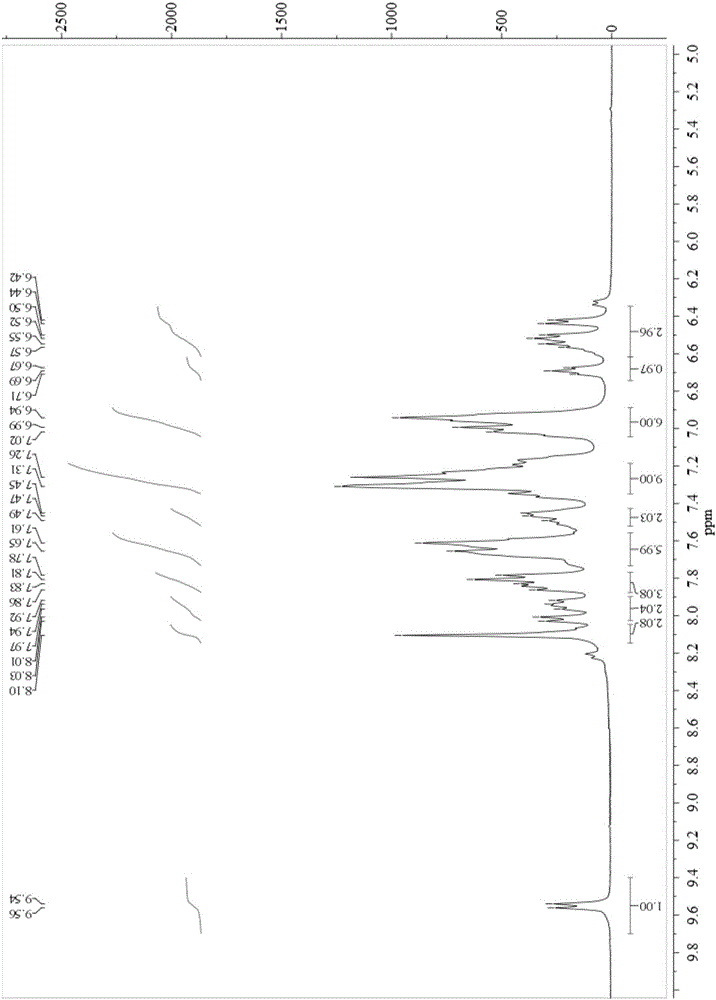
图1为配合物1的1H NMR核磁图;
图2为配合物2的1H NMR核磁图;
图3为配合物3的1H NMR核磁图;
图4为配合物1在乙腈溶液中的紫外吸收光谱和室温、低温及薄膜的发射光谱;
图5为配合物2在乙腈溶液中的紫外吸收光谱和室温、低温及薄膜的发射光谱;
图6为配合物3在乙腈溶液中的紫外吸收光谱和室温、低温及薄膜的发射光谱;
图7为配合物1、2和3的热重分析(TGA)曲线;
图8为电致发光器件ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物1(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)D1、ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物2(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)D2、ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物3(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)D3的EL光谱;
图9为电致发光器件D1、D2和D3的发光亮度-电压-电流密度特性曲线;
图10为电致发光器件D1、D2和D3的电流效率-电流密度-外量子效率曲线。
具体实施方式
本发明的配合物可按照如下方程式进行合成:
核磁共振氢谱是在Bruker DPX 400(400MHz)核磁共振仪上测定;ESI-MS质谱是在Esquire-LC_00136质谱仪上测定,C、H、N的元素分析是在Exeter Analytical CE-440型元素分析仪上测定;紫外吸收光谱是在Agilent Cary 60紫外-可见上测定;荧光发射光谱是在Hitachi F-7000荧光光谱仪上测定;OLED器件是在真空压力低于3×10-5Pa的真空度镀膜机中蒸镀沉积制备,器件的光电特性曲线是在Keithley Source 2400半导体性能测试系统测定,电致发光光谱是在Photo Research PR650光谱仪上测定。
实施例1
配合物1的合成:
在无水无氧条件下,将双-[7,8-苯并喹啉]合二氯化铱(0.50克,0.43毫摩)和2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑的钾盐(0.43克,1.08毫摩)溶于15毫升乙二醇单乙醚中,于120℃反应12小时,过滤,重结晶,最后通过柱层层析提纯得到配合物二[7,8-苯并喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱(1)。
配合物2的合成:
在无水无氧条件下,将双-[2-苯基喹啉]合二氯化铱(0.50克,0.39毫摩)和2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑的钾盐(0.39克,0.98毫摩)溶于15毫升乙二醇单乙醚中,于130℃反应12小时,过滤,重结晶,最后通过柱层层析提纯得到配合物二[2-苯基喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱(2)。
配合物3的合成:
在无水无氧条件下,将双-[1-苯基异喹啉]合二氯化铱(0.50克,0.39毫摩)和2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑的钾盐(0.39克,0.98毫摩)溶于15毫升乙二醇单乙醚中,于150℃反应12小时,过滤,重结晶,最后通过柱层层析提纯得到配合物二[1-苯基异喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱(3)。
配合物1、2和3经1H NMR、质谱、元素分析进行了验证,结果表明结构正确,数据如下:
配合物1,产率为63%:
1H NMR(400MHz,CDCl3,δ):9.52(d,J=4.0Hz,1H),8.54(d,J=4.0Hz,1H),8.16(d,J=8.0Hz,1H),7.89(d,J=8.0Hz,1H),7.81–7.74(m,4H),7.59–7.49(m,4H),7.44–7.34(m,6H),7.21–7.06(m,7H),6.94–6.87(m,2H),6.81(s,2H),6.38(d,J=8.0Hz,1H),6.17(d,J=8.0Hz,1H)。
MS(ESI-MS)[m/z]:m/z 909.8(M+H)+。
元素分析结果:计算值:C(%):60.78,H(%):3.44,N(%):7.70。
实测值:C(%):60.75,H(%):3.46,N(%):7.72。
配合物2,产率为68%:
1H NMR(400MHz,CDCl3,δ):9.56(d,J=8.0Hz,1H),8.10(s,2H),8.03–7.92(m,2H),7.86–7.78(m,3H),7.66–7.59(m,6H),7.49–7.45(m,2H),7.35–7.17(m,9H),7.02–6.94(m,6H),6.69(t,J=8.0Hz,1H),6.57–6.42(m,3H)。
MS(ESI-MS)[m/z]:m/z 961.8(M+H)+。
元素分析结果:计算值:C(%):62.49,H(%):3.67,N(%):7.29。
实测值:C(%):62.53,H(%):3.65,N(%):7.27。
配合物3,产率为73%:
1H NMR(400MHz,CDCl3,δ):9.25(s,1H),8.99(s,2H),8.25–8.19(m,3H),7.87–7.57(m,11H),7.41–7.36(m,8H),6.97–6.84(m,4H),6.75–6.41(m,6H)。
MS(ESI-MS)[m/z]:m/z 961.8(M+H)+。
元素分析结果:计算值:C(%):62.49,H(%):3.67,N(%):7.29。
实测值:C(%):62.47,H(%):3.68,N(%):7.31。
实施例2
配合物1、2和3的紫外吸收光谱、发射光谱和其它表征:
将配合物1、2和3分别溶于乙腈中(10-5M),除氧,在Agilent Cary 60和Hitachi F-7000光谱仪上分别测定其吸收和发射光谱(含低温和薄膜发射光谱):
配合物的吸收光谱和发射光谱的峰位置分别为:
二[7,8-苯并喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱(1):
λabs,max,nm 255,299,441(见附图4)
λem,max,nm 539(乙腈溶液),518(低温),534(薄膜)(见附图4)
二[2-苯基喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱(2):
λabs,max,nm 270,337,453(见附图5)
λem,max,nm 592(乙腈溶液),575(低温),583(薄膜)(见附图5)
二[1-苯基异喹啉]-{2-(N-二苯基次磷酰)-氨基-5-苯基-[1,3,4]-噁二唑}合铱(3):
λabs,max,nm 292,390,462(见附图6)
λem,max,nm 614(乙腈溶液),605(低温),610(薄膜)(见附图6)
为客观评价配合物1、2和3的光谱性质,分别测定了这三个配合物在溶液和掺杂PMMA薄膜(5wt%)中的发光量子产率,配合物1、2和3的发光量子产率分别为21%(1,乙腈溶液)、6%(2,乙腈溶液)、6%(3,乙腈溶液)、35%(1,PMMA薄膜)、37%(2,PMMA薄膜)和18%(3,PMMA薄膜)。
配合物1、2和3都有较好的热稳定性,热分解初始温度(对应于5%的质量损失):
Td,℃ 367(1);356(2);333(3)(见附图7)
实施例3
以配合物1、2和3为发光中心的有机电致发光器件OLEDs的制备:
器件制备仪器:德国Mbraun公司MB-MO-SE1型真空热蒸发镀膜设备;测试仪器:Keithley Source 2400,Photo Research PR650光谱仪。
器件的结构为:
D1:ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物1(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)D1
D2:ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物2(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)D2
D3:ITO/HIO2(25nm)/NPB(5nm)/TCTA(10nm)/mCP:配合物3(20nm)/TPBI(40nm)/Liq(1nm)/Al(100nm)D3
器件的电流效率(cd/A)由器件的I-V和L-V特性得到:
ηc=L/I (1)
器件的功率效率可由下式计算得到:
ηp=π×S×L/(I×V) (2)
其中,L为发光亮度,I为电流密度,S为发光面积,V为加载电致发光器件两端的电压。
以15Ω/sq的ITO玻璃为衬底,先用玻璃清洗剂清洗干净,再用去离子水,丙酮各超声3遍,经紫外-臭氧下处理15分钟后,进行有机层蒸镀。首先,25nm的空穴注入材料HIO2被沉积到ITO玻璃基底上,然后沉积5nm的空穴传输材料NPB,然后沉积10nm的激子阻挡材料TCTA,随后客体材料和主体材料以共蒸的形式形成20nm的发光层,接下来是40nm的电子传输和空穴阻挡材料TPBI,1nm的电子注入材料Liq和100nm的阴极铝。在材料蒸镀沉积时,真空室的压力低于3×10-5Pa。器件的发光亮度-电压-电流密度、电流效率-电流密度-外量子效率曲线是在Keithley Source 2400半导体性能测试系统测定,电致发光光谱是在Photo Research PR650光谱仪上测定。所有测量均在室温大气下进行,器件的主要性能如下:
电致发光器件D1、D2和D3的EL光谱:
λEL,max,nm 534(D1);588(D2);613((D3)(见附图8)
电致发光器件D1、D2和D3的发光亮度-电压-电流密度曲线:
最大发光亮度Lmax,cd/m2:29800(D1,9V);19340(D2,13.5V);7007(D3,13.5V)(见附图9)
电致发光器件D1、D2和D3的电流效率-电流密度-外量子效率曲线:
最大电流效率ηc,max,cd/A:60.77(D1);25.99(D2);7.56(D3),且器件表现为较弱的效率滚降现象,见图10。
- 还没有人留言评论。精彩留言会获得点赞!