一种环金属铱配合物及其制备方法与作为电化学发光标记物的应用与流程
本发明属于生物分析领域,具体涉及生物分子的电化学发光标记物和生物分子的电化学发光分析检测。
背景技术:
在生物分析检测领域,电化学发光免疫是继放射免疫、酶免疫、荧光免疫、化学发光免疫之后的新一代微量标记免疫分析技术,其具有检测灵敏度高(可以达到飞摩尔量级)、选择性好、检测时间快、线性范围宽等优点,被广泛用于基础医学、临床应用、食品安全等领域。目前成功商业化的电化学发光标记分子主要是三联吡啶钌衍生物,但其具有明显的缺点,如:发光颜色比较单一,发光量子效率低,发光波长很难通过分子结构进行调节等弊端。
现有技术公开了一些环金属化合物,理论上具有较好的电化学发光性能;但是不具反应性,无法与生物分子形成偶联,导致其无法实际应用,即不可用于生物分子标记,而且现有应用体系都为有机相,由于生物分子在有机相中生物活性丧失,进一步导致其无法实际应用;另外化合物本身的电性质对其能否作为生物分子标记物以及标记效果有着极其重要的影响。因此,开发新型高效的生物分子电化学发光信号标记物对于电化学发光生物分析检测技术的发展具有重要意义。
技术实现要素:
本发明的目的是提供一类新型高效的生物分子电化学发光信号标记物。该类电化学发光标记物的可以通过配体结构的调节实现电化学发光发射波长从红到蓝绿色的调节,同时具有较高的电化学发光效率,利用其所修饰的羧酸官能团可以快速实现与生物分子的共价偶联,化学稳定性高。本发明可以有效克服传统电化学发光标记物的弊端,具有巨大的市场应用前景和良好的经济社会效益。
为达到上述发明目的,本发明采用的技术方案是:一种环金属铱配合物,其化学结构式为:
其中二齿配体为第一配体,为第二配体;所述环金属铱配合物属于离子型化合物,X为对离子,可以为氯离子(Cl-)、六氟磷酸根(PF6-)和四氟硼酸根(BF4-)。
上述技术方案中,二齿配体为第一配体,可以为以下化学结构式中的一种:
其中,R为氢、甲基、甲氧基、硝基、氰基或者卤素等;
为第二配体,可以选用以下化学结构式中的一种:
其中,n可以是1到9中的任意整数。
优选的,所述环金属铱配合物的结构式为以下化学结构式中的一种:
本发明公开了上述环金属铱配合物作为生物分子电化学发光信号标记物的应用;所述生物分子包括蛋白质分子、多肽分子、脱氧核糖核酸分子、核糖核酸分子;本发明的环金属铱配合物修饰的羧基具有良好的反应性,可以与生物分子进行反应,从而实现对生物分子的标记。现有技术认为烷烃链段由于具有碳碳键,比苯环或者杂环更容易振动,从而更容易吸收电能而转化为热能,从而削弱电化学发光,这对生物标记十分不利;本发明通过结构设计,创造性的公开的环金属铱配合物带有羧基基团,实现与生物分子的共价偶联,化学稳定性高;同时具有电化学发光效率高的优点,参见本发明实施例,同等条件下,本发明金属铱标记物的电化学发光强度明显强于现有成熟的金属钌配合物;取得了意想不到的技术效果。
本发明还公开了上述环金属铱配合物在生物分子电化学发光检测中的应用;所述生物分子,可以是蛋白质分子,多肽,脱氧核糖核酸(DNA),核糖核酸(RNA)等,优选蛋白质分子和脱氧核糖核酸分子。环金属铱配合物标记生物分子后,电化学发光信号产生体系为水相体系,可以是含有电化学发光共反应物的磷酸盐缓冲溶液,三羟甲基氨基甲烷(Tris)缓冲溶液和4-羟乙基哌嗪乙磺酸(HEPES)缓冲溶液。水相体系是实现生物分子电化学检测的必要条件,现有环金属化合物大都在有机相具有良好的电化学发光,但是无法应用在水相;本发明的环金属铱配合物克服了现有技术难题,可以在缓冲液中对生物分子进行标记,然后在水相体系中进行电化学检测,具有优于现有成熟的金属钌配合物的技术效果。
本发明还公开了一种生物分子电化学发光检测的方法,以上述环金属铱配合物为标记物,以叔胺化合物为共反应物,在水相体系中,实现生物分子电化学发光检测;所述生物分子包括蛋白质分子、多肽分子、脱氧核糖核酸分子、核糖核酸分子。环金属铱配合物在生物分析标记中,羧酸基团需要首先转化为-N-羟基琥珀酰亚胺酯,然后再与生物分子进行偶联。所述水相体系为磷酸盐缓冲溶液体系、三羟甲基氨基甲烷缓冲溶液体系或者4-羟乙基哌嗪乙磺酸缓冲溶液体系;所述叔胺类化合物为正-三丙胺或者三乙胺。
本发明还公开了上述环金属铱配合物的制备方法,包括以下步骤:
(1)环醚溶剂中,化合物A与强碱反应;然后加入溴腈化合物,继续反应,得到化合物B;然后将化合物B在浓酸中回流反应,得到二齿配体;所述化合物A的化学结构式为以下化学结构式的一种:
(2)三氯化铱与二齿配体在乙二醇乙醚中反应,得到过渡金属氯桥化合物,然后将氯桥化合物与二齿配体反应得到环金属铱配合物。
上述技术方案中,所述强碱为二异丙基氨基锂;所述溴腈化合物为溴丙腈或者溴丁腈;所述浓酸为浓硫酸。
由于上述技术方案运用,本发明与现有技术相比具有下列优点:
(1)本发明公开的环金属铱配合物作为电化学发光标记物,波长可以实现从蓝光到红光的调节,有利于开发多通道的免疫分析设备和产品,提高检测的效率;并且电化学发光效率高;克服了现有技术中电化学发光化合物发光颜色单一,发光量子效率低的缺陷;具有巨大的市场应用前景和良好的经济社会效益;
(2)本发明公开的环金属铱配合物具有羧酸基团,可以快速实现与生物分子的共价偶联,且化学稳定性高;尤其是解决了现有含烷烃链段的化合物由于碳碳键振动导致的电化学性能削弱的问题,得到的环金属铱配合物具有高效的电化学发光效率;
(3)本发明公开的环金属铱配合物适用水相体系,在生物分析检测领常采用的水相体系中,与传统的电化学发光标记物相比,同等条件下,具有更高的电化学发光信号强度,从而可以提高对生物分子的检测灵敏度,而且适于实际应用。
附图说明
图1为实施例一电化学发光标记物的电化学发光信号图;
图2为实施例三电化学发光标记物的电化学发光信号图;
图3为实施例七电化学发光标记物的电化学发光信号图;
图4为实施例八DNA的电化学发光信号图;
图5为实施例九BSA的电化学发光信号图;
图6为含不同主配体结构标记物的电化学发光波长图。
具体实施方式
实施例一 所用的电化学发光标记物的结构式如下:
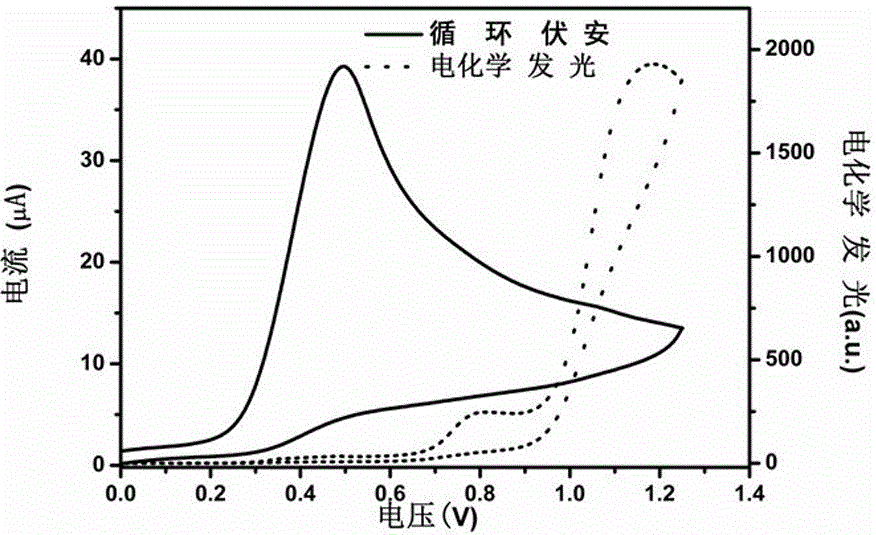
以正-三丙胺为共反应物,在PBS缓冲溶液中,在电极上施加0 V-1 V-0 V循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,随着正-三丙胺被氧化,检测到最大的电化学发光信号,如图1所示。
实施例二 所用的电化学发光标记物的结构式如下:
以正-三丙胺为共反应物,在PBS缓冲溶液中,在电极上施加0 V-1 V-0 V循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,随着正-三丙胺被氧化,检测到最大的电化学发光信号。
实施例三 所用的电化学发光标记物的结构式如下:
以三乙胺为共反应物,在Tris缓冲溶液中,在电极上施加0 V-1.2 V-0 V循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,随着共反应物被氧化,检测到最大的电化学发光信号,如附图2所示。
实施例四 所用的电化学发光标记物的结构式如下:
以正-三丙胺为共反应物,在Tris缓冲溶液中,在电极上施加0 V-1.3 V-0 V循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,检测到的电化学发光信号逐渐增强。
实施例五 所用的电化学发光标记物的结构式如下:
以正-三丙胺为共反应物,在PBS缓冲溶液中,在电极上施加0 V-1.1 V-0 V循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,检测到的电化学发光信号逐渐增强。
实施例六 所用的电化学发光标记物的结构式如下:
以三乙胺为共反应物,在PBS缓冲溶液中,在电极上施加0 V-1.05 V-0 V循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,检测到的电化学发光信号逐渐增强。
实施例七 所用的电化学发光标记物的结构式如下:
以正-三丙胺为共反应物,在Tris缓冲溶液中,在电极上施加0 V-1.1 V-0 V线性循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,检测到的电化学发光信号逐渐增强,如附图3所示。
实施例八 所用的电化学发光标记物的结构式如下:
将标记物在DMF中利用二环己基碳二亚胺(DCC),N-羟基琥珀酰亚胺(NHS)作为催化剂,将羧酸基团转化为NHS酯,然后与末端氨基修饰的DNA链在PBS溶液中反应1小时,然后将DNA链进行纯化,将未标记的DNA链和其中游离的标记物分子除去,然后用正-三丙胺做为共反应剂,在PBS缓冲溶液中,对工作电极施加0 V-1.5 V-0 V线性循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,检测到的电化学发光信号逐渐增强,如附图4所示。
实施例九 所用的电化学发光标记物的结构式如下:
将标记物在DMF中利用二环己基碳二亚胺(DCC),N-羟基琥珀酰亚胺(NHS)作为催化剂,将羧酸基团转化为NHS酯,然后与牛血清蛋白(BSA)在PBS溶液中反应2小时后,利用透析方法将游离的小分子标记物去除。然后在含有正-三丙胺共反应物的Tris缓冲溶液中,对工作电极施加0 V-1.2 V-0 V线性循环伏安扫描,随着扫描电位逐渐增加,电极表面电流逐渐增大,检测到的电化学发光信号逐渐增强。同等实验条件下,利用传统的电化学发光标记物三联吡啶钌衍生进行BSA蛋白标记,发现同等浓度被标记的BSA蛋白,金属铱标记物的电化学发光强度明显强于传统的金属钌配合物,如附图5所示,同等浓度下,Ir标记的蛋白质具有更强的电化学发光信号。
实施例十
化合物1溶于无水四氢呋喃中,-78℃低温条件下与LDA强碱反应2小时后,加入溴丙腈,继续在室温条件下反应18小时,经柱层析纯化得到中间产物化合物2,然后将中间产物在浓硫酸条件下加热回流24小时,得到目标产物3。其他对应的含不同亚甲基的羧酸,可以通过溴代氰基类化合物的碳原子数进行改变。
化合物4溶于无水四氢呋喃中,-78℃低温条件下与LDA强碱反应2小时后,加入溴丁腈,继续在室温条件下反应24小时,经柱层析纯化得到中间产物化合物5,然后将中间产物在浓硫酸条件下加热回流12小时,得到目标产物6。
三氯化铱与二齿配体在乙二醇乙醚溶剂中反应24小时后,得到过渡金属氯桥化合物,然后进一步将氯桥化合物与二齿配体化合物反应得到最终的电化学发光标记物环金属铱配合物。
本发明所涉及的其他电化学发光化合物的合成路线与此类似。
图6是含不同主配体结构标记物的电化学发光波长,图中从左到右曲线代表的环金属铱配合物的化学结构式依次如下:
。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种羰基锝-99m标记的美法仑配合物及其制备方法与用图
- 一种2-羰基-3-苯基丙酸苯甲酰腙二苯基锡配合物及其制备方法和应用
- 一种新型金属有机配合物纤维及其衍生多孔碳纤维的制备方法
- 包含新的配体结构的金属配合物的制作方法
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯苄基)锡配合物及其制备方法和应用
- 一种2-羰基-2-苯基乙酸苯甲酰腙二丁基锡配合物及其制备方法和应用
- 一种2-羰基-3-苯基丙酸水杨酰腙二苯基锡配合物及其制备方法和应用