基于脂质体膜的金属有机骨架材料的表面功能化修饰方法与流程
本发明属于纳米材料制备技术领域,尤其涉及一种基于脂质体膜的金属有机骨架材料的表面功能化修饰方法。
背景技术:
金属有机骨架材料(metal-organicframeworks,mofs)是以无机金属离子簇为配位节点,以有机配体为连接单元,通过配位作用形成的一种三维孔状材料。由于具有多样的结构组成,较高的比表面积,可调节的孔径大小,可调控的形貌尺寸以及生物相容性好等特点,mofs材料被广泛应用于药物递送、分子成像、生物传感等生物医学领域。从金属有机骨架材料的结构特点来看,其表面存在未配位的金属离子结合位点和未连接的有机配体连接节点,因此在进行生物医学应用时必须对其表面进行功能化修饰以提高其生物稳定性与生物安全性。传统的表面功能化修饰方法如在mofs材料表面连接聚乙二醇,虽然在一定程度上可以提高mofs材料的生物稳定性与生物安全性,但仍然无法满足mofs材料在应用于生物医学领域时对生物稳定性的需求,同时也无法改善mofs材料在递送一些成像分子或治疗药物时会出现提早释放的现状,因此,迫切需要发展一种新的、功能化更强的修饰手段,不仅可以显著提高mofs材料的生物稳定性,还能在此基础赋予mofs材料一些新的功能,例如实现递送分子的可控释放等。
综上所述,现有技术存在的问题是:传统mofs材料表面功能化修饰方法存在应用于生物医学领域时对生物稳定性差,无法改善mofs材料在递送成像分子或治疗药物时出现提早释放。
技术实现要素:
针对现有技术存在的问题,本发明提供了一种基于脂质体膜的金属有机骨架材料的表面功能化修饰方法。
本发明是这样实现的,一种基于脂质体膜的金属有机骨架材料的表面功能化修饰方法,所述基于脂质体膜的金属有机骨架材料的表面功能化修饰方法使脂质体膜通过自主融合的方式包覆至mofs材料表面;
所述基于脂质体膜的金属有机骨架材料的表面功能化修饰方法具体包括:
(1)选择相互匹配的mofs材料与脂质体,使脂质体膜与mofs材料表面裸露的活性位点之间存在静电吸附或共价连接作用;
(2)将相应的mofs材料与脂质体溶液直接混合,进行融合包覆,离心洗涤得到最终产物;
(3)进行质量评价与性能评估,确定通过静电吸附或共价连接,脂质体成功实现对mofs材料的包覆,完成对mofs材料的表面功能化修饰。
进一步,所述mofs材料包括uio系列、mil系列以及zif系列;
所述uio系列包括uio-66、uio-66-nh2;
所述mil系列包括mil-100、mil-101-nh2、mil-88a、mil-53;
所述zif系列包括zif-8、zif-7、zif-11、zif-5。
进一步,所述脂质体包括阴离子脂质体、中性脂质体、阳离子脂质体;
所述阴离子脂质体为dopa脂质体、dppa脂质体、dopg脂质体、dops脂质体;
所述中性脂质体为dopc脂质体、dppc脂质体;
所述阳离子脂质体为dotap脂质体。
进一步,所述脂质体的合成原料包括1,2-二油酰基卵磷脂dopc,1,2-二油酰磷脂酸dopa,2-二油酰基羟丙基-3-n,n,n-三甲铵氯dotap,二油酰基磷脂酰丝氨酸dops,二油酰磷脂酰甘油dopg,二油酰基磷脂酰乙醇胺-聚乙二醇18:1peg-2000pe,胆固醇chol。
进一步,所述mofs材料的合成方法包括以下步骤:
(1)用溶剂热合成法合成uio系列与zif系列,称取金属离子与有机配体将其超声分散在相应的溶剂中,加入调节剂后转移至水热釜中反应,离心超声再分散,用溶剂洗涤几次得到最终产物;
(2)用微波合成法合成mil系列,称取金属离子与有机配体将其超声分散在相应的溶剂中,转移至微波反应瓶中,在微波合成仪上进行反应,转速离心超声再分散,用溶剂洗涤几次得到最终产物。
进一步,合成uio-66时金属离子为无水氯化锆,称取的量为10~30mg;有机配体为对苯二甲酸,称取的量为30~80mg;反应溶剂为n,n-二甲基甲酰胺,量取的量为3~6ml;调节剂为冰醋酸,量取的量为0.2~1ml;反应温度为60~110℃;反应时间为10~24h;离心过程转速为10000rpm,时间为10min,洗涤试剂为n,n-二甲基甲酰胺与无水乙醇,洗涤次数为2~5次;
合成zif-8时金属离子为六水合硝酸锌,称取的量为1.0~3.5g;有机配体为二甲基咪唑,称取的量为4.3~7.2g;反应溶剂为甲醇,量取的量为50~200ml;反应温度为15~60℃;反应时间为0.5~12h;离心过程转速为12000rpm,时间为15min,洗涤试剂为甲醇与无水乙醇,洗涤次数为2~5次。
进一步,合成mil-100时金属离子为六水合三氯化铁,称取的量为1.0~3.5g;有机配体为均苯三甲酸,称取的量为4.3~7.2g;反应溶剂为水与乙醇的混合溶液,量取的量为50~200ml;反应温度为15~60℃;反应时间为0.5~12h;离心过程转速为12000rpm,时间为15min,洗涤试剂为甲醇与无水乙醇,洗涤次数为2~5次;
合成mil-101-nh2时金属离子为六水合三氯化铁,称取的量为1.0~3.5g;有机配体为邻氨基对苯二甲酸,称取的量为4.3~7.2g;反应溶剂为,量取的量为50~200ml;反应温度为15~60℃;反应时间为0.5~12h;离心过程转速为12000rpm,时间为15min,洗涤试剂为甲醇与无水乙醇,洗涤次数为2~5次。
进一步,所述脂质体的合成方法具体包括:
用挤出器挤出法制备脂质体,按质量配比称取各种脂质2.5~10mg,溶于2~4ml氯仿和甲醇的混合溶液中,体积比为4:1,溶解完全混合均匀后在水浴中用旋转蒸发仪将有机溶剂抽干,室温下真空干燥过夜;加入4~6mlhepes缓冲液室温水化1~2h,然后借助脂质体挤出器挤出21次,得到最终的脂质体。
进一步,所述合成dopa脂质体时质量配比为dopa:chol:18:1peg-2000pe=15:6:1,水浴温度为35℃;
合成dopc脂质体时脂质的质量配比为dopc:chol:18:1peg-2000pe=15:6:1,水浴温度为25℃;
合成dotap脂质体时脂质的质量配比为dopa:chol:18:1peg-2000pe=15:6:1,水浴温度为40℃。
本发明的另一目的在于提供一种由所述基于脂质体膜的金属有机骨架材料的表面功能化修饰方法修饰的金属有机骨架材料。
本发明的优点及积极效果为:选择带电性质不同,亲水端基团不同的脂质体分别探究与mofs材料的自主融合情况,发现利用脂质体膜与mofs材料表面间的静电吸附或共价连接均可实现脂质双分子层在mofs材料表面的自主融合,使其成功包覆在mofs材料的表面,使得修饰后的mofs材料具有超高的生物稳定性。脂质体作为一个综合的运输载体已经被广泛应用于成像造影剂、小分子药物、生物分子(包括寡核苷酸、多肽、蛋白以及气体分子)的运输与递送。脂质体主要由磷脂双分子层构成,与细胞膜的组成类似,因此具有较高的生物稳定性与生物安全性。和传统的mofs材料表面修饰方法相比,利用生物相容性较好的脂质体膜对mofs表面进行包覆是一种新颖的表面功能化修饰手段,具有良好的应用前景。
本发明通过脂质体膜与mofs材料表面间的静电吸附或共价连接,均可实现脂质体膜在mofs材料表面的自主融合与包覆,完成对mofs材料的表面功能化修饰。该发明构建了一种操作简单、绿色高效的针对mofs材料进行表面功能化修饰的新方法,为mofs材料的表面功能化修饰提供了一种新的思路;在提高mofs材料生物稳定性与生物安全性的同时还赋予了mofs材料一些新的功能,例如亲疏水性药物分子的共同递送、递送分子的可控释放等,对其在生物医学领域方面的应用具有重要意义。此外也为mofs材料与脂质体的有机结合提供了一个新的范例,利用脂质体膜完成表面修饰的mofs材料将有望同时拥有两种材料的优越性,极大地拓宽了mofs材料与脂质体在生物医学领域等方面的应用前景。
本发明所提供的针对mofs材料进行表面功能化修饰的方法主要是以静电吸附或共价作用作为驱动力,来实现脂质体膜在mofs材料表面的自主融合,完成对mofs材料的表面功能化修饰,在目前的期刊与论文中未出现公开过。相比于传统的聚乙二醇修饰方法可以提供更高的生物稳定性;操作简便,仅通过mofs材料与脂质体直接混合便可一步实现,且修饰效率也相对较高;不需要使用有机溶剂或其他有毒试剂,符合绿色合成的理念;将脂质体与mofs材料有机地结合在一起,在应用于药物递送、分子影像等方面时有望赋予mofs材料新的功能。
附图说明
图1是本发明实施例提供的基于脂质体膜的金属有机骨架材料的表面功能化修饰方法流程图。
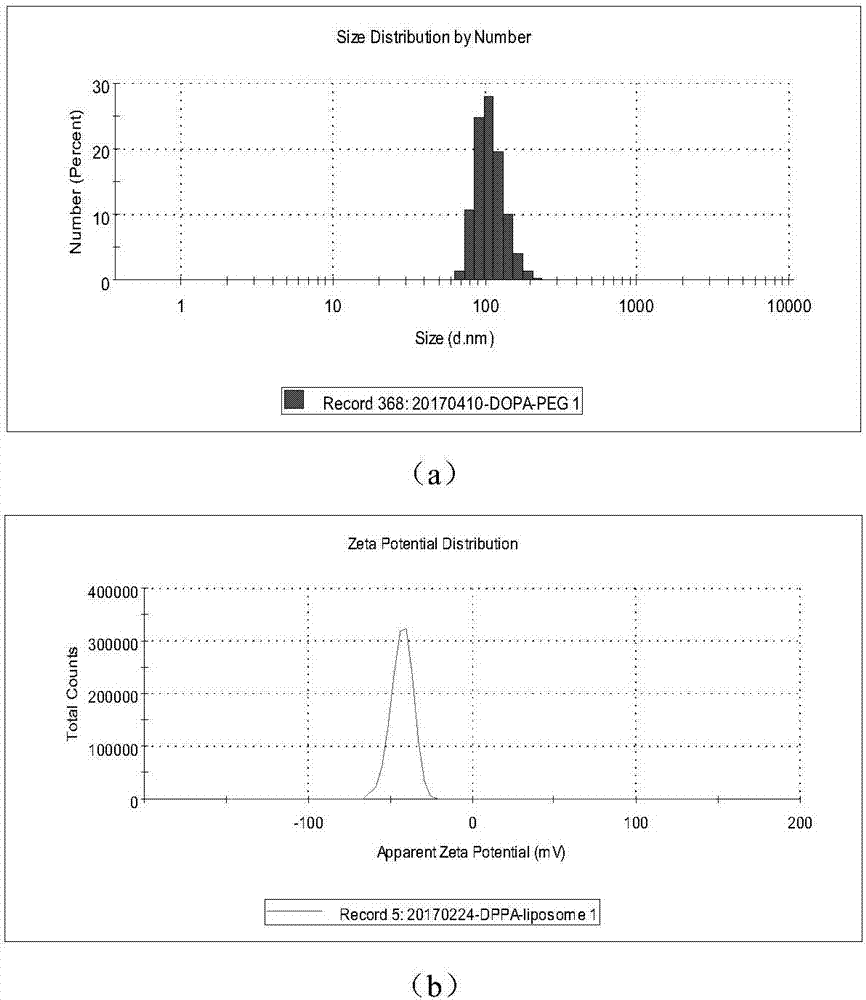
图2是本发明实施例提供的实施例1中制备的dopa脂质体的水合动力学尺寸图与电位图;
图中:(a)dopa脂质体的水合动力学尺寸图;(b)为dopa脂质体的电位图。
图3是本发明实施例提供的实施例2中制备的uio-66的扫描电镜照片与透射电镜照片示意图;
图中:(a)为水合动力学直径为230nm的uio-66的扫描电镜照片;(b)为水合动力学直径为150nm的uio-66的透射电镜照片。
图4是本发明实施例提供的实施例3中制备的uio-66@dopa的水合动力学尺寸图与电位图;
图中:(a)uio-66@dopa的水合动力学尺寸图;(b)为uio-66@dopa的电位图。
图5是本发明实施例提供的实施例中dopa脂质体、uio-66、uio-66@dopa的红外图谱示意图;
其中1为dopa脂质体的红外图谱;2为uio-66的红外图谱;3为uio-66@dopa的红外图谱。
图6是本发明实施例提供的实施例6中uio-66-peg与uio-66@dopa在pbs缓冲液中的稳定性变化图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
mofs材料固有的结构特点决定了其表面必然存在许多裸露的活性位点,即未配位的金属离子结合位点和未连接的有机配体连接单元,具有良好的表面修饰基础。脂质体膜内外表面裸露的基团具有多样性与可选择性。通过合理地选择mofs材料与脂质体的种类来进行匹配,利用脂质体膜与mofs材料表面的相互作用如范德华力、静电作用或共价作用,将实现脂质体膜在mofs材料表面上的有效包覆,极大地提高mofs材料的生物稳定性。
下面结合附图对本发明的应用原理作详细的描述。
如图1所示,本发明实施例提供的基于脂质体膜的金属有机骨架材料的表面功能化修饰方法包括以下步骤:
s101:称取0.1~2.0mg的mofs材料,超声分散在1~4ml的相应的脂质体溶液中,浓度为2.5mg/ml,在室温下静置0.5~2h,每隔5~15min用移液枪进行吹打混匀几次,反应结束后用去离子水洗涤三次,洗去未包覆在mofs材料表面的脂质体,最终得到表面包覆一层脂质双分子层的mofs材料,即mofs@liposome;
s102:利用脂质体成功包覆在mofs材料的表面,完成表面功能化修饰后mofs材料相比于未修饰前的mofs材料由于脂质体膜较薄,约为5~10nm,水动力学尺寸基本不变,表面电位与所选用的包覆的脂质体的电位基本相同。
本发明的mofs材料包括uio系列、mil系列以及zif系列等。
其中uio系列包括uio-66、uio-66-nh2等;mil系列包括mil-100、mil-101-nh2、mil-88a、mil-53等;zif系列包括zif-8、zif-7、zif-11、zif-5等。
本发明的脂质体包括阴离子脂质体如dopa脂质体、dppa脂质体、dopg脂质体、dops脂质体等;中性脂质体如dopc脂质体、dppc脂质体等;阳离子脂质体如dotap脂质体等。
脂质体的合成原料包括1,2-二油酰基卵磷脂(dopc),1,2-二油酰磷脂酸(dopa),2-二油酰基羟丙基-3-n,n,n-三甲铵氯(dotap),二油酰基磷脂酰丝氨酸(dops),二油酰磷脂酰甘油(dopg),二油酰基磷脂酰乙醇胺-聚乙二醇(18:1peg-2000pe),胆固醇(chol)
mofs材料优先选择uio-66、mil-100、mil-101-nh2。
脂质体优先选择dopa脂质体、dotap脂质体、dopc脂质体。
本发明mofs材料的合成方法具体包括:
(1)用溶剂热合成法合成uio系列与zif系列,具体步骤为称取适量的金属离子与有机配体将其超声分散在相应的溶剂中,加入适量的调节剂后转移至水热釜中,在一定温度下反应一段时间,以一定的转速离心超声再分散,用合适的溶剂洗涤几次得到最终产物。
上述步骤中合成uio-66时金属离子为无水氯化锆,称取的量为10~30mg,具体可为25mg;有机配体为对苯二甲酸,称取的量为30~80mg,具体可为40mg;反应溶剂为n,n-二甲基甲酰胺,量取的量为3~6ml,具体可为4ml;调节剂为冰醋酸,量取的量为0.2~1ml,具体可为0.5ml;反应温度为60~110℃,具体可为80℃;反应时间为10~24h,具体可为15h;离心过程转速为10000rpm,时间为10min,洗涤试剂为n,n-二甲基甲酰胺与无水乙醇,洗涤次数为2~5次,具体可为3次。
合成zif-8时金属离子为六水合硝酸锌,称取的量为1.0~3.5g,具体可为2.0g;有机配体为二甲基咪唑,称取的量为4.3~7.2g,具体可为5.6g;反应溶剂为甲醇,量取的量为50~200ml,具体可为130ml;反应温度为15~60℃,具体可为25℃;反应时间为0.5~12h,具体可为1h;离心过程转速为12000rpm,时间为15min,洗涤试剂为甲醇与无水乙醇,洗涤次数为2~5次,具体可为3次。
(2)用微波合成法合成mil系列,具体步骤为称取适量的金属离子与有机配体将其超声分散在相应的溶剂中,将其转移至微波反应瓶中,在微波合成仪上设置相应的反应条件进行反应,以一定的转速离心超声再分散,用合适的溶剂洗涤几次得到最终产物。
合成mil-100时金属离子为六水合三氯化铁,称取的量为1.0~3.5g,具体可为2.0g;有机配体为均苯三甲酸,称取的量为4.3~7.2g,具体可为5.6g;反应溶剂为水与乙醇的混合溶液,量取的量为50~200ml,具体可为130ml;反应温度为15~60℃,具体可为25℃;反应时间为0.5~12h,具体可为1h;离心过程转速为12000rpm,时间为15min,洗涤试剂为甲醇与无水乙醇,洗涤次数为2~5次,具体可为3次。
合成mil-101-nh2时金属离子为六水合三氯化铁,称取的量为1.0~3.5g,具体可为2.0g;有机配体为邻氨基对苯二甲酸,称取的量为4.3~7.2g,具体可为5.6g;反应溶剂为,量取的量为50~200ml,具体可为130ml;反应温度为15~60℃,具体可为25℃;反应时间为0.5~12h,具体可为1h;离心过程转速为12000rpm,时间为15min,洗涤试剂为甲醇与无水乙醇,洗涤次数为2~5次,具体可为3次。
本发明脂质体的合成方法具体包括:
用挤出器挤出法制备脂质体,具体的实现过程为按一定的质量配比称取各种脂质2.5~10mg,溶于2~4ml氯仿和甲醇的混合溶液中,体积比为4:1,溶解完全混合均匀后在一定的水浴温度下用旋转蒸发仪将有机溶剂抽干,室温下真空干燥过夜。加入4~6mlhepes缓冲液(100mmnacl,10mmhepes,ph7.4)室温水化1~2h,然后借助脂质体挤出器挤出21次,得到最终的脂质体。
合成dopa脂质体时质量配比为dopa:chol:18:1peg-2000pe=15:6:1,水浴温度为35℃。
合成dopc脂质体时脂质的质量配比为dopc:chol:18:1peg-2000pe=15:6:1,水浴温度为25℃。
合成dotap脂质体时脂质的质量配比为dopa:chol:18:1peg-2000pe=15:6:1,水浴温度为40℃。
本发明脂质体包覆mofs材料的合成方法具体包括:
称取0.1~2.0mg的mofs材料,超声分散在1~4ml的相应的脂质体溶液中,浓度为2.5mg/ml,在室温下静置0.5~2h,每隔5~15min用移液枪进行吹打混匀几次,反应结束后用去离子水洗涤三次,洗去未包覆在mofs材料表面的脂质体,最终得到表面包覆一层脂质双分子层的mofs材料,即mofs@liposome。
上述方法合成dopa包覆uio-66即uio-66@dopa时,称取的uio-66的量为0.1~0.5mg,具体可为0.3mg,dopa脂质体的量为1ml,其中uio-66@dopa形成的驱动力为uio-66表面裸露的锆离子与dopa脂质体膜上的磷酸基团的共价作用。
合成dotap包覆mil-100即mil-100@dopa时,称取的mil-100上的量为0.5~2.0mg,具体可为1mg,dotap脂质体的量为1ml,其中mil-100@dotap形成的驱动力为mil-100表面裸露的羧酸根与dotap脂质体膜上的胆碱基团的静电作用。
最终利用脂质体成功包覆在mofs材料的表面,完成表面功能化修饰后mofs材料相比于未修饰前的mofs材料由于脂质体膜较薄,约为5~10nm,水动力学尺寸基本不变,表面电位与所选用的包覆的脂质体的电位基本相同,生物稳定性能得到了较大的提升。
下面结合具体实施例对本发明的应用原理作进一步的描述。
实施例1
脂质体的制备
dopa脂质体的制备
称取5mg脂质,其中各种脂质组分的质量比为dopa/chol/18:1peg-2000pe=15:6:1,溶于2ml氯仿和甲醇的混合溶液中(体积比是4:1),溶解完全混合均匀后在35℃水浴条件下用旋转蒸发仪将有机溶剂抽干,室温下真空干燥过夜。加入4mlhepes缓冲液(100mmnacl,10mmhepes,ph7.4)室温水化1~2h,然后借助脂质体挤出器挤出21次,得到尺寸为120~130nm的dopa脂质体。如图2(a)所示为dopa脂质体的水合动力学直径图,如图2(b)所示为dopa脂质体的电位图。
dopc脂质体的制备
称取5mg脂质,其中各种脂质组分的质量比为dopc/chol/18:1peg-2000pe=15:6:1,溶于2ml氯仿和甲醇的混合溶液中(体积比是4:1),溶解完全混合均匀后在25℃水浴条件下用旋转蒸发仪将有机溶剂抽干,室温下真空干燥过夜。加入4mlhepes缓冲液(100mmnacl,10mmhepes,ph7.4)室温水化1~2h,然后借助脂质体挤出器挤出21次,得到水动力学尺寸为120~130nm的dopc脂质体。
dotap脂质体的制备
称取5mg脂质,其中各种脂质组分的质量比为dotap/chol/18:1peg-2000pe=15:6:1,溶于2ml氯仿和甲醇的混合溶液中(体积比是4/1),溶解完全混合均匀后在40℃水浴条件下用旋转蒸发仪将有机溶剂抽干,室温下真空干燥过夜。加入4mlhepes缓冲液(100mmnacl,10mmhepes,ph7.4)室温水化1~2h,然后借助脂质体挤出器挤出21次,得到水动力学尺寸为120~130nm的dotap脂质体。
实施例2
mofs材料的制备
uio-66的制备
称取15.5mg氯化锆(zrcl4),50mg对苯二甲酸(h2bdc),溶于4mln,n-二甲基甲酰胺(dmf)中,溶解完全,加入0.3~0.9ml的乙酸,转移至水热反应釜,在90~100℃下反应18h,得到的溶液在10000rpm转速下离心10min,通过超声分散与离心用乙醇与水分别洗两遍,得到uio-66,其水动力学尺寸为140~900nm。如图3(a)所示为水动力学直径为230nm的uio-66的扫描电镜图,如图3(b)所示为水动力学直径为150nm的uio-66的透射电镜图。
mil-100的制备
称取486mg的六水合氯化铁(fecl3·6h2o),140mg的均苯二甲酸(h3btc),溶于5ml水与乙醇的混合溶液中,其中水与乙醇的体积比为1:0.25~4,溶解完全,转移至10ml的微波反应瓶中,在微波合成仪中130℃反应5min,得到的溶液在10500rpm转速下离心20min,通过超声分散与离心用乙醇洗三遍,得到mil-100,水动力学尺寸为150~500nm。
mil-101-nh2的制备
称取27mg的六水合氯化铁(fecl3·h2o),18.1mg的邻氨基对苯二甲酸(h2bdc-nh2),溶于5ml水与乙醇的混合溶液中,其中水与乙醇的体积比为1:0.25~4,溶解完全,转移至10ml的微波反应瓶中,在微波合成仪中60℃反应5min,得到的溶液在10500rpm转速下离心10min,通过超声分散与离心用乙醇洗三遍,得到mil-101-nh2,水动力学尺寸为170~500nm。
zif-8的制备
称取2.4g的六水和硝酸锌(zn(no3)2·6h2o),5.28g的甲基咪唑(2-mim),溶于100ml的甲醇中,溶解完全,转移至250ml的圆底烧瓶中进行磁力搅拌,在室温下反应30~90min,得到的溶液在7800rpm转速下离心1.5h,通过超声分散与离心用甲醇洗三遍,得到zif-8,水动力学尺寸为100~300nm。
zif-7
称取136mg六水和硝酸锌(zn(no3)2·6h2o),120mg苯并咪唑(bim),溶于5mln,n-二甲基甲酰胺(dmf)中,溶解完全,转移至25ml的圆底烧瓶中进行磁力搅拌,在室温下反应10~30min,得到的溶液在7800rpm转速下离心1.5h,通过超声分散与离心分别用dmf与甲醇洗三遍,得到zif~7,水动力学尺寸为300~600nm。
实施例3
dopa脂质体共价连接包覆uio-66
dopa脂质亲水端末尾为磷酸基团,可以与uio-66表面裸露的未配位的锆离子结合位点进行共价连接,使dopa脂质体自主融合在uio-66的表面,从而完成对uio-66的包覆,记为uio-66@dopa,具体的实现过程为将1mg的uio-66超声分散在1ml浓度为1mg/ml的dopa脂质体溶液中,在室温下静置1h,每个10min用移液枪进行吹打混匀几次,反应结束后用去离子水洗涤三次,洗去未包覆在uio-66表面的dopa脂质体,最终得到表面包覆一层dopa脂质双分层的uio-66,即uio-66@dopa。如图4(a)所示为uio-66@dopa的水动力学直径图,如图4(b)所示为uio-66@dopa的电位图,为了进一步证明uio-66@dopa的成功构建,对uio-66、dopa脂质体、uio-66@dopa分别进行红外图谱的测定,如图5所示,测定结果显示uio-66@dopa同时拥有uio-66与dopa脂质体特有的官能团,证明dopa脂质体成功包覆在uio-66的表面。
实施例4
dopc脂质体静电吸附包覆mil-100
dopc脂质亲水端末尾为磷酸基团与胆碱基团,尽管为最终的带电性质为中性,但其疏水端末尾带正电的胆碱基团仍然可以与mil-100表面裸露的未连接的羧酸根离子连接位点进行静电吸附,使dopc脂质体自主融合在mil-100的表面,从而完成对mil-100的包覆,记为mil-100@dopc,具体的实现过程为将0.5mg的mil-100超声分散在1ml浓度为1mg/ml的dopc脂质体溶液中,在室温下静置1h,每个10min用移液枪进行吹打混匀几次,反应结束后用去离子水洗涤三次,洗去未包覆在mil-100表面的dopc脂质体,最终得到表面包覆一层dopc脂质双分层的mil-100,即mil-100@dopc。
实施例5
uio-66-peg的制备
nh2-peg可以通过共价作用与uio-66表面裸露的羧酸根连接位点相连接,并且用edc/nhs进行活化可以提高反应效率,具体的实现过程为称取0.5mg的uio-66,将其超声分散在1ml的nh2-peg-5000dmf溶液中,其中nh2-peg-5000dmf溶液浓度为3~5mg/ml,再加入5mg的nhs与1mg的edc,在25℃的条件下涡旋震荡12h,10000rpm离心10min,通过离心再分散的方式用去离子水洗涤3次,洗去未反应的nh2-peg、edc、nhs,最终得到表面连接上peg的uio-66,即uio-66-peg。
实施例6
uio-66-peg与uio-66@dopa的生物稳定性测定
利用pbs缓冲液与10%胎牛血清培养基在体外模拟生理环境,依据uio-66-peg与uio-66@dopa-liposome在相应的条件下水动力学直径尺寸的变化来对对其生物稳定性进行测定。设置四组分别进行测定,具体的实现过程如下:
组1为uio-66-peg在pbs缓冲液中稳定性测定:取0.5mg的uio-66-peg超声分散至5ml的pbs缓冲液中,置于37℃水浴中,分别在0h、0.5h、1h、2h、4h、8h、16h、32h、48h时进行水动力学尺寸的测定。
组2为uio-66-peg在10%胎牛血清培养基中稳定性测定:取0.5mg的uio-66-peg超声分散至5ml的10%胎牛血清培养基中,置于37℃水浴中,分别在0h、0.5h、1h、2h、4h、8h、16h、32h、48h时进行水动力学尺寸的测定。
组3为uio-66@dopa在pbs缓冲液中稳定性测定:取0.5mg的uio-66@dopa超声分散至5ml的pbs缓冲液中,置于37℃水浴中,分别在0h、0.5h、1h、2h、4h、8h、16h、32h、48h时进行水动力学尺寸的测定。
组4为uio-66@dopa在10%胎牛血清培养基中稳定性测定:取0.5mg的uio-66-peg超声分散至5ml的10%胎牛血清培养基中,置于37℃水浴中,分别在0h、0.5h、1h、2h、4h、8h、16h、32h、48h时进行水动力学尺寸的测定。结果显示uio-66@dopa在pbs缓冲液与10%胎牛血清培养基中的稳定性均要优于uio-66-peg,在pbs缓冲液中的稳定性测试结果如图6所示。
实施例7
mil-100@dopc用于亲疏水药物的共同递送
由于mil-100更倾向于运载亲水性药物,因此无法实现具有联合化疗效果的亲水性药物吉西他滨与疏水性药物紫杉醇的共同运载。mil-100@dopc则可以通过在其表面的脂质双分子层中插入紫杉醇来实现化疗药物吉西他滨与紫杉醇的共同递送。具体的实现过程为包括以下步骤:
(1)dopc脂质体运载紫杉醇:将1~5mg的紫杉醇与5~10mg的脂质共同分散在4ml氯仿与甲醇的混合溶液中,其中各种脂质组分的质量比为dopc:chol:pe~peg2000=15:6:1,氯仿与甲醇的体积比为4:1,25℃水浴旋蒸除去所用的有机溶剂,室温真空干燥12h后加入4mlhepes缓冲液,水化1~2h,用装有孔径为100nm挤出膜的挤出器挤出21次得到。
(2)mil-100运载吉西他滨:将1mg的mil-100超声分散在1ml浓度为2mg/ml的吉西他滨水溶液中,在室温下缓慢搅拌12h,反应结束后用去离子水洗涤三次,洗去未载入在mil-100腔体内的吉西他滨,最终得到运载有吉西他滨的mil-100。
将1mg的装载吉西他滨的mil-100超声分散在1ml浓度为1mg/ml的运载了紫杉醇的dopc脂质体溶液中,在室温下静置1h,每个10min用移液枪进行吹打混匀几次,反应结束后用去离子水洗涤三次,洗去未包覆在mil-100表面的dopc脂质体,最终得到运载了具有联合化疗效果的吉西他滨与紫杉醇的mil-100@dopc。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!