转基因构建物及其应用的制作方法
本发明属于基因工程领域,特别涉及一种转基因构建物及在制备p53基因全部阅读框定点敲出和双报告基因组织特异性表达分子影像小鼠中的应用。
背景技术:
将外源基因导入动物机体或敲出动物机体内源性基因或深入探讨基因功能需要转基因技术,特别地,探讨基因在动物机体的功能或改良生物性状,研究基因在疾病特别是肿瘤发生、发展和转移中的作用,需要采用转基因过表达或基因敲出技术。
疾病相关动物模型在阐明基因功能、人类重大疾病发病机制及新药研发等方面为人类健康与进步做出了巨大贡献。肿瘤发生的组织特异性和器官选择性相关机制研究是肿瘤研究领域的一大难题,目前符合临床发病规律的动物模型十分匮乏。
荧光蛋白的发现和分子影像学技术的出现为肿瘤的早期发现,研究肿瘤发生、发展和转移相关机制提供了重要手段和新途径。
抑癌基因p53在大多数的人类癌症如白血病,淋巴瘤,肉瘤,脑瘤,乳腺癌,胃肠道癌及肺癌等癌症中常呈失活现象,其突变可高达50%以上,因此它是人类恶性肿瘤中最常见的基因改变。p53基因的产物具有调节细胞周期和细胞凋亡,参与dna修复,调控肿瘤新生血管形成等功能。p53发生缺陷的肿瘤细胞不易发生凋亡,维持了肿瘤细胞的生存,也增加了对化疗药物和放射治疗的耐药性和不敏感性。因此,p53的生物学调控在调节正常细胞的生长,控制肿瘤形成及癌症的治疗中起着至关重要的作用。目前还没有p53基因全部阅读框敲出和双荧光报告基因组织特异性表达基因编辑小鼠,因此本领域迫切需要建立和开发该种工具小鼠。
技术实现要素:
为解决上述技术问题,本发明提供了一种转基因构建物及制备p53定点敲出和双荧光报告基因组织特异性表达分子影像小鼠中的应用。
本发明的另一个目的是将基因敲出、基因敲入技术同分子影像学技术有效结合起来,利用小动物成像系统等多种分子影像学技术手段实时无创伤检测转基因动物由于p53基因缺失对基因转录调控机制的影响;通过构建mcherry组织特异性表达模型,为研究肿瘤发生的器官选择性分子机制及观察肿瘤治疗药物的器官靶向性建立一个生物模型。
本发明的第一方面,提供一种转基因构建物,所述的基因构建物包括包括小鼠位于第11号染色体上的mtrp53基因(genbankaccessionnumber:nm_0116403;ensembl:
ensmusg00000059552)的从位于第二外显子的翻译起始密码子atg开始上游4953bp的5′同源臂基因组dna片段、一种基因表达盒以及从位于第11外显子的翻译终止密码子tga开始下游2487bp的3′同源臂基因组dna片段。该基因构建物可与小鼠mtrp53基因的位于第二外显子atg开始上游4953bp和位于第11外显子的终止密码子tga开始下游2487bp基因组dna片段发生同源重组,将该区间基因组dna片段敲出,同时将相等长度的基因表达盒dna片段定点敲入到该区间。
其中所述5′同源臂基因组dna片段的核苷酸序列如seqidno.:1所示;其中所述3′同源臂基因组dna片段的核苷酸序列如seqidno.:2所示。
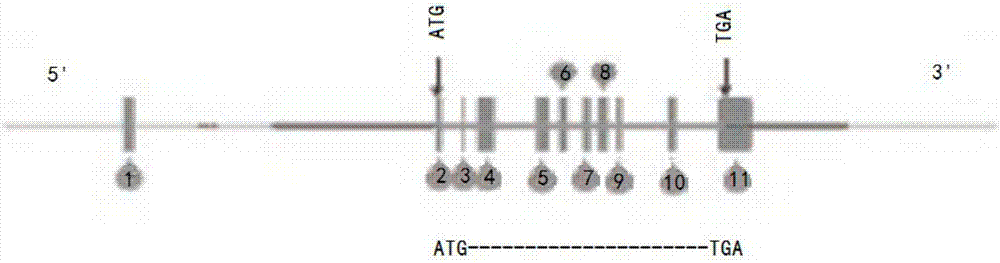
上述方案中,所述的转基因构建物将与小鼠位于第11号染色体上的mtrp53基因的第二外显子的翻译起始密码子atg开始上游4953bp和位于第11外显子的翻译终止密码子tga开始下游2487bp基因组dna片段发生同源重组,将该区间基因组dna片段敲出,同时将相等长度的基因表达盒定点敲入到该区间,该转基因构建物敲出基因组区间结构框架如图二所示。
本发明的第二方面,提供了一种基因表达盒,所述的基因表达盒从5’至3’依次具有下述元件:萤火虫荧光素酶(fireflyluciferase)表达基因、polya元件、cag启动子元件、loxp位点序列、polya元件、loxp位点序列、mcherry荧光蛋白表达基因、polya元件、neomycin抗性表达基因。
上述方案中,所述的基因表达盒中的萤火虫荧光素酶(fireflyluciferase)表达基因受小鼠p53基因内源性启动子调控。
上述方案中,所述的基因表达盒中的mcherry荧光蛋白表达基因的表达受基因表达盒中的cag启动子元件调节。所述的基因表达盒中cag启动子元件和mcherry荧光蛋白表达基因之间存在loxp位点序列、polya元件、loxp位点序列,使得正常情况下cag启动子无法启动mcherry荧光蛋白表达基因的表达。所述的基因表达盒中的cag启动子启动mcherry荧光蛋白表达基因的表达受外源性cre转基因的表达所调控。
本发明的第三方面,提供了一种载体,所述载体含有或本发明第一方面所述的转基因构建物,或第二方面所述的基因表达盒。
在另一个优选例中,所述载体选自细菌质粒、噬菌体、酵母质粒、病毒、或动物细胞载体、穿梭载体。
本发明的第四方面,提供了一种宿主细胞,所述宿主细胞含有发明第三方面所述的载体,或其染色体整合有第一方面所述的转基因构建物,或其染色体整合有发明第二方面所述的基因表达盒。
在另一个优选例中,所述宿主细胞的染色体含有一个或多个拷贝的发明第一方面所述的转基因构建物或第二方面所述的基因表达盒。
在另一个优选例中,所述的宿主细胞选自下组:原核细胞(如大肠杆菌),低等真核细胞(如酵母细胞)、或高等真核细胞(如动物细胞)。
在另一个优选例中,所述的宿主细胞为小鼠细胞。
本发明的第五方面,提供了第一方面所述的转基因构建物,或发明第二方面所述基因表达盒的用途,用于精确定点敲出小鼠mtrp53基因的从位于第二外显子atg开始到位于第11外显子的终止密码子tga之间的基因组dna片段,同时将相等长度的基因表达盒dna片段定点敲入到该区间。构建p53基因全部阅读框定点敲出和双报告基因组织特异性表达分子影像基因编辑小鼠模型。
本发明的第六方面,提供了一种制备转基因动物的方法,包括如下步骤:
(1)提供转基因动物细胞,所述动物细胞含有发明第三方面所述的载体,或其染色体整合有第一方面所述的转基因构建物,或其染色体上整合有第二方面所述的基因表达盒;
(2)将步骤(1)所述的转基因细胞再生为动物,从而获得转基因动物。
本发明的第七方面,提供了一种转基因动物的用途,所述的转基因动物是用发明第六方面所述的方法制备的,所述的转基因动物用于研究p53基因敲出对小鼠遗传、发育、生长和肿瘤发生及转移的影响,研究肿瘤发生的器官选择性分子机制以及观察肿瘤治疗药物的组织器官靶向性、特异性。
本发明的第八方面,提供了一种在动物中表达萤火虫荧光素酶(fireflyluciferase)基因的方法,包括如下步骤:
(1)提供发明第一方面所述的转基因构建物;
(2)将步骤(1)中的转基因构建物导入动物细胞,获得转化的动物细胞;
(3)将步骤(2)中转化的动物细胞再生为动物,从而使萤火虫荧光素酶(fireflyluciferase)基因的表达受p53基因内源性启动子调控。
本发明的第九方面,提供一种在动物中表达mcherry荧光蛋白基因的方法,包括如下步骤:
(1)提供发明第一方面所述的转基因构建物;
(2)将步骤(1)中的转基因构建物导入动物细胞,获得转化的动物细胞;
(3)将步骤(2)中转化的动物细胞再生为动物,从而使mcherry荧光蛋白基因的表达受权利要求3所述基因表达盒中的cag启动子的调控。
本发明的第十方面,提供一种在动物中可以选择性表达mcherry荧光蛋白基因的方法,包括如下步骤:
(1)提供发明第一方面所述的转基因构建物;
(2)将步骤(1)中的转基因构建物导入动物细胞,获得转化的动物细胞;
(3)将步骤(2)中转化的动物细胞再生为动物,从而使mcherry荧光蛋白基因的表达受权利要求3所述基因表达盒中的cag启动子的调控;
(4)将步骤(3)中获得的动物与组织特性表达cre转基因动物交配,从而获得组织特异性表达mcherry荧光蛋白基因的子代动物。
(5)将步骤(3)中获得的小鼠与其他具有不同遗传背景的小鼠交配,从而获得含有发明第一方面所述的转基因构建物和相关遗传背景的杂交小鼠。
应理解,在本发明范围内,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一赘述。
通过上述技术方案,本发明提供的转基因构建物及在制备p53基因全部阅读框定点敲出和双报告基因组织特异性表达分子影像小鼠中的应用,特别是首次建立了双荧光报告基因组织器官特异性表达技术平台;同时本发明结合分子影像学技术手段可为研究p53基因敲出对小鼠遗传、发育、生长和肿瘤发生及转移的影响,研究肿瘤发生的器官选择性分子机制以及观察肿瘤治疗药物的组织器官靶向性、特异性提供了一个重要模型。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
图1显示基因构建物结构框架图。
图2显示基因组dna敲出片段结构框架图。
图3显示基因表达盒结构框架图。
图4显示转基因构建物的结构及限制性内切酶图谱。
图5显示基因编辑小鼠制备技术路线与方案。
图6显示es细胞打靶鉴定结果(pcr)。
图7显示es细胞打靶验证结果(southernblot)。
图8显示flox阳性小鼠与flox杂合阳性/tek-cre杂合阳性小鼠鉴定策略与结果(基因组dna-pcr法)。
图9显示flox阳性杂合小鼠和flox阳性杂合/tek-cre阳性杂合小鼠luc和mcherry表达(小动物成像系统)检测结果。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。
本发明经过广泛而深入的研究,首次建立了p53基因全部阅读框定点敲出和双荧光报告基因组织特异性表达分子影像小鼠模型。具体地,发明人利用同源重组-es打靶技术将小鼠位于第11号染色体上的mtrp53基因(genbankaccessionnumber:nm_0116403;ensembl:ensmusg00000059552)的从位于第二外显子的翻译起始密码子atg到位于第11外显子的翻译终止密码子tga之间的基因组dna片段定点敲出,同时将fireflyluciferase-polya-cag-loxp-stop-loxp-mcherry-polya基因表达cassette定点敲入到上述位点,使得fireflyluciferase基因的表达受内源性p53基因启动子的调控,mcherry的表达依赖于cre重组酶的组织特异性表达。应用这种基因编辑技术制备基因定点敲出/敲入分子影像小鼠模型,可利用小动物成像系统实时无创伤监测由于p53基因敲出对小鼠遗传、发育、生长和肿瘤发生及转移的影响,通过构建mcherry组织特异性表达模型,可以研究肿瘤发生的器官选择性分子机制及观察肿瘤治疗药物的器官靶向性,同时本发明建立了进行肿瘤药物治疗药效学评价一个重要模型。
用于制备重组打靶载体的方法是本领域技术人员所熟知的,表达载体可以是细菌质粒,噬菌体,酵母质粒,哺乳动物细胞病毒或其他载体。
用于制备重组打靶载体5′端和3′端同源臂的基因组dna片段是以c57bl/6j文库中的bac克隆rp-23-5i20和rp23-51o12为模版由pcr扩增产生。打靶载体设计方案与结构如图1,2,3,4所示。
c57bl/6es细胞做为基因打靶的宿主细胞,基因打靶载体经过限制性内切酶线性化消化后,利用电穿孔的方法将线性化的打靶载体导入es细胞。经过neomycin药物初筛获得neomycin抗性细胞,最后经过pcr筛选与southernblot验证,从96个耐药克隆中发现了14个基因打靶成功的克隆,其中6个克隆经由southernblot的方法得到了验证。基因打靶技术路线与方案如图5所示,利用同源重组-es打靶技术将小鼠位于第11号染色体上的mtrp53基因(genbankaccessionnumber:nm_0116403;ensembl:ensmusg00000059552)从位于第二外显子的翻译起始密码子atg到位于第11外显子的翻译终止密码子tga之间的基因组dna片段定点敲出,同时将fireflyluciferase-polya-cag-loxp-stop-loxp-mcherry-polya基因表达cassette定点敲入到上述位点。重组打靶载体5′端和3′端同源臂的基因组dna片段是以c57bl/6j文库中的bac克隆rp-23-5i20和rp23-51o12为模版由pcr扩增产生。基因表达盒中cag启动子元件和mcherry荧光蛋白基因之间存在loxp位点序列、polya元件、loxp位点序列,基因表达盒中的mcherry的表达依赖于cre重组酶的特异性表达。
用于pcr筛选基因打靶成功的es细胞的策略与方法如图6,7所示。针对打靶载体上三个不同的部位设计了三组pcr引物,用于es细胞阳性克隆的筛查。具体地,如图6所示,pcr筛选基因打靶成功的es细胞的策略与方法如所示(a)。针对打靶载体上三个不同的部位设计了三组pcr引物,用于es细胞阳性克隆的筛查(b)。
针对打靶载体region1设计的pcr引物,阳性es克隆dna经pcr扩增后的片段长度是3.7kb,野生型es细胞dna经pcr扩增后的无扩增片段(c上图)。
针对打靶载体region2设计的pcr引物,阳性es克隆dna经pcr扩增后的片段长度是3.1kb,野生型es细胞dna经pcr扩增后的无扩增片段(c中图)。
针对打靶载体region3设计的pcr引物,阳性es克隆dna经pcr扩增后的片段长度是2.8kb,野生型es细胞dna经pcr扩增后的无扩增片段(c下图)。
6个经过pcr鉴定阳性的es克隆,利用southernblot的方法进行了验证(方法如a所示)。具体地,如图7所示,分别提取es阳性细胞基因组dna,基因组dna分别利用aflii或xmai和asci限制性酶消化,分别利用含有neo基因5′端和3′端的dna片段为探针,进行southernblot分析,最后验证得到6个阳性克隆。利用xmai和asci限制性酶消化后得到的阳性片段程度为15.4kb,利用aflii限制性酶消化后得到的阳性片段程度为19.8kb(图b)。
将阳性es克隆1h1,1d3和1b3利用显微注射方法注射入代孕鼠囊胚,制备嵌合体小鼠,并通过毛色判断嵌合体(黑白相间),黑色占的比例越大,嵌合率越高。
去除抗性基因neo:具体地,嵌合体小鼠和全身都表达flp酶的小鼠杂交,把打靶载体中的neo基因去除,获得f1代杂合子。
f1代阳性杂合子小鼠通过pcr鉴定得到。鉴定方案和所用引物(如图a,b所示)。具体地,如图8所示,提取f1小鼠鼠尾基因组dna,利用图8所示的策略和pcr引物进行鉴定,阳性杂合小鼠基因组dna经pcr扩增后的dna片段长度为508bp。与cre转基因小鼠交配后得到的携带有cre转基因的阳性小鼠基因组dna经pcr扩增后的dna片段程度为413bp。
获得f1代杂合子后,挑选一对雌雄杂合子f1代ko小鼠进行自交,通过pcr鉴定筛选得到去除frt两个位点片段纯合子的f2代ko小鼠(命名:geneflox/flox)。备注:geneflox/flox两条染色体上都带有loxp-gene-loxp的模型。
mcherry组织特异性表达小鼠模型的建立。f2代杂合或纯合阳性小鼠分别与组织特异性cre小鼠(tek-cre和pvillin-cre转基因小鼠)杂交,pcr鉴定,得到敲除p53基因并表达cre的杂合小鼠。鉴定方法如图9所示。
利用多模式小动物成像系统检测f3代p53基因敲出杂合子和组织特异性表达cre杂合小鼠体内luciferase和mcherry的表达。如图9所示,具体地,小动物成像系统显示在f3代p53基因敲出杂合小鼠体内有荧光素酶的广泛表达a(左上图flox+/-小鼠),同时该小鼠体内没有mcherry的表达水平极低b(左下图flox+/-小鼠)。在f3代p53基因敲出杂合/tek-cre杂和小鼠体内有荧光素酶的广泛表达c(右上图flox+/-/tek-cre+/-小鼠),同时该小鼠体内由于有cre转基因的组织特异性表达导致mcherry有较高水平的组织特异性表达d(右下图flox+/-/tek-cre+/-小鼠),提示该小鼠模型的建立成功。
在本发明的一个具体实例中,该模型小鼠的p53基因从位于第二外显子atg开始到位于第十一外显子的tga为止全部被敲出掉。
在本发明的一个具体实例中,该模型小鼠在以上相应位点定点敲入与敲出片段等长的基因表达盒。如图3所示,该基因表达盒包含萤火虫荧光素酶(fireflyluciferase)表达基因ⅰ、polya元件ⅱ、cag启动子元件ⅲ、loxp位点序列ⅳ、polya元件ⅴ、loxp位点序列ⅵ、mcherry荧光蛋白表达基因ⅶ、polya元件ⅷ、neomycin抗性表达基因ⅸ(ffluc-polya-cag-loxp-stop-loxp-mcherry-polya)。
在本发明的一个具体实例中,所述的基因表达盒中的萤火虫荧光素酶(fireflyluciferase)基因的表达受小鼠位于p53第二外显子atg上游内源性启动子元件调控。
在本发明的一个具体实例中,所述的基因表达盒中的mcherry荧光蛋白基因的表达受基因表达盒中的cag启动子元件调节。
在本发明的一个具体实例中,所述的基因表达盒中cag启动子元件和mcherry荧光蛋白基因之间存在loxp位点序列、polya元件、loxp位点序列,使得正常情况下cag启动子无法启动mcherry荧光蛋白基因的表达。
在本发明的一个具体实例中,所述的基因表达盒中的mcherry的表达依赖于cre重组酶的组织特异性表达。
本发明的主要优点如下:
本发明首次将小鼠位于第11号染色体上的mtrp53基因(genbankaccessionnumber:nm_0116403;ensembl:ensmusg00000059552)的从位于第二外显子的翻译起始密码子atg到位于第11外显子的翻译终止密码子tga之间的基因组dna片段定点全部敲出。
同时将同等长度的fireflyluciferase-polya-cag-loxp-stop-loxp-mcherry-polya基因表达cassette定点敲入到上述位点,使得fireflyluciferase基因的表达受内源性p53基因启动子的调控,mcherry的表达依赖于cre重组酶的组织特异性表达。
本发明将基因敲出、基因敲入技术同分子影像学技术有效结合起来,首次建立p53基因敲出-双荧光报告基因定点敲入基因编辑分子影像小鼠模型,可利用小动物成像系统实时无创伤监测由于p53基因敲出对小鼠遗传、发育、生长和肿瘤发生及转移的影响。
通过构建mcherry组织特异性表达模型,可以研究肿瘤发生的器官选择性分子机制及观察肿瘤治疗药物的器官靶向性。
同时本发明建立了可用于肿瘤药物治疗药效学评价一个重要模型。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种新型转基因复合物及其制备方法与应用
- 一种动物饲料添加剂的制备方法
- Cmp?乙酰神经氨酸羟化酶打靶载体、载体转导的异种移植用转基因动物及其制造方法
- 辛酰化Ghrelin在促进牛卵母细胞体外成熟中的应用
- 一种添加中草药的实验动物颗粒垫料及其制备方法
- 一种靶向Mesothelin的复制缺陷性重组慢病毒CAR-T转基因载体及其构建方法和应用
- 一种靶向cd123的复制缺陷性重组慢病毒car-t转基因载体及其构建方法和应用
- 一种靶向cd30的复制缺陷性重组慢病毒car-t转基因载体及其构建方法和应用
- 一种靶向cd22的复制缺陷性重组慢病毒car-t转基因载体及其构建方法和应用
- 可穿戴式多通道无线皮层脑电检测装置的制造方法