一种对羟基苯甲醛的合成方法与流程
本发明属于精细化工技术领域,具体涉及一种对羟基苯甲醛的合成方法。
背景技术:
对羟基苯甲醛是一种重要的香料、高分子材料、医药、农药中间体。以对羟基苯甲醛为原料,可以合成合成香兰素、乙基香兰素、洋茉莉醛、丁香醛、茴香醛和复盆子酮等多种香料。在农药领域,对羟基苯甲醛主要用来合成除草剂溴苯腈和羟敌草腈等。在医药领域,对羟基苯甲醛主要于羟氨苄青霉素(阿莫西林)、抗菌增效剂甲氧苄胺嘧啶(tmp)、3,4,5-三甲氧基苯甲醛、对羟基甘氨酸、羟氨苄头孢霉素、人造天麻、杜鹃素、艾思洛儿等药物的合成。此外对羟基苯甲醛还用于杀菌剂、照相乳化剂、镀镍光泽剂、液晶等产品的合成。
对羟基苯甲醛的生产有多条工艺路线,目前工业生产主要有苯酚法、对甲酚催化氧化法、对硝基甲苯法等原料路线。(1)苯酚法又分为reimer-tiemann反应、gattermann反应、苯酚-三氯乙醛路线、苯酚-乙醛酸路线、苯酚-甲醛路线等多种合成工艺路线。苯酚法的工艺特点是原料易得,制造工艺较简单,但收率偏低,成本较高。(2)对硝基甲苯法生产对羟基苯甲醛的工艺过程分氧化还原、重氮化和水解三步进行。产物经提取、纯化、干燥得对羟基苯甲醛产品,收率能达到90%以上。此工艺的优点是原料价格便宜,但缺点是工艺路线长,设备庞大,且中间产物对氨基苯甲醛有毒,重氮化反应温度低,冷冻条件高,不利于工业化应用。(3)对甲酚催化氧化法,该工艺在催化剂作用下,用空气或氧直接氧化对甲酚合成对羟基苯甲醛。该工艺以对甲酚为原料,要用含钴催化剂,原料短缺,催化剂制备工艺复杂,成本较高。
技术实现要素:
本发明所要解决的技术问题在于针对上述现有技术的不足,提供了一种对羟基苯甲醛的合成方法。该合成方法原料转化率高,产品收率高,是一种以苯酚和甲酸为原料制备对羟基苯甲醛的新方法,即在催化剂a的作用下,苯酚和甲酸发生酯化反应,生成甲酸苯酯,甲酸苯酯在催化剂b的作用下,甲酸苯酯发生fries重排反应,合成对羟基苯甲醛,经重结晶提纯后,产品纯度可达到99%以上,产品综合收率最高可达到95%。该方法原料来源广泛,反应过程简单,所用催化剂廉价易得,对生产设备无苛刻要求,设备投资小,便于工业化应用。
为解决上述技术问题,本发明采用的技术方案是:一种对羟基苯甲醛的合成方法,其特征在于,该方法为:以苯酚和甲酸为原料,在催化剂a的作用下,发生酯化反应,生成甲酸苯酯;再在催化剂b的作用下,所述甲酸苯酯发生fries重排反应,生成对羟基苯甲醛。
上述的一种对羟基苯甲醛的合成方法,其特征在于,该方法包括以下步骤:
步骤一、将苯酚、甲酸、催化剂a和有机溶剂均加到反应器中,加热至100℃~180℃进行酯化反应,酯化反应结束后,停止加热并降温至40℃,得到反应液;然后对反应液进行蒸馏,收集171℃~173℃的馏分,得到甲酸苯酯;
步骤二、将催化剂b、有机溶剂和步骤一中得到的甲酸苯酯均加到反应器中,加热至100℃~180℃进行fries重排反应,fries重排反应结束后停止加热并降温至40℃,然后向所述反应器中加入去离子水或稀盐酸,边搅拌边加热至60℃,恒温搅拌30min,然后分液得到有机相,最后对得到的有机相进行浓缩,除去有机溶剂后得到浅黄色固体;
步骤三、用乙醇和水的混合溶剂对步骤二中得到的浅黄色固体进行重结晶,得到白色的对羟基苯甲醛晶体。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤一中所述苯酚、甲酸和催化剂a的摩尔比为1:(1.5~2.5):(0.05~0.1),所述有机溶剂的加入体积等于苯酚的摩尔量,所述有机溶剂加入体积的单位为l,所述苯酚摩尔量的单位为mol。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤一和步骤二中所述有机溶剂均为甲苯、对二甲苯、氯苯或邻二氯苯。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤一中所述催化剂a为浓硫酸、苯磺酸、对甲苯磺酸或对氯苯磺酸,所述浓硫酸的质量浓度98%。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤二中所述甲酸苯酯和催化剂b的摩尔比为1:(1~3),所述有机溶剂的加入体积等于苯酚的摩尔量,所述有机溶剂加入体积的单位为l,所述苯酚摩尔量的单位为mol。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤二中所述催化剂b为三氯化铝或氯化锌。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤二中所述去离子水或稀盐酸的加入体积均为有机溶剂体积的1/3~1倍,所述稀盐酸的质量分数为10%。
上述的一种对羟基苯甲醛的合成方法,其特征在于,步骤三中所述混合溶剂中乙醇和水的体积比为1:1。
本发明与现有技术相比具有以下优点:
1、本发明是以苯酚、甲酸为原料,通过酯化反应和fries重排反应合成对羟基苯甲醛,原料易得,反应时间短,操作步骤简单,反应条件温和,反应过程安全,对设备要求低,相对其他对羟基苯甲醛的合成方法,本发明更适用于工业生产对羟基苯甲醛。
2、本发明在合成过程中没有副产物产生,后期处理过程中分离提纯方法简单,降低了后期处理的成本,提高了目标产物对羟基苯甲醛的收率和纯度,最佳反应条件下的综合收率在95%以上,纯度在99%以上,解决了现有工艺中因产物分离和提纯困难而造成的对羟基苯甲醛收率偏低和成本较高的问题。
下面通过附图和实施例对本发明的技术方案作进一步的详细说明。
附图说明
图1是本发明的反应路线图。
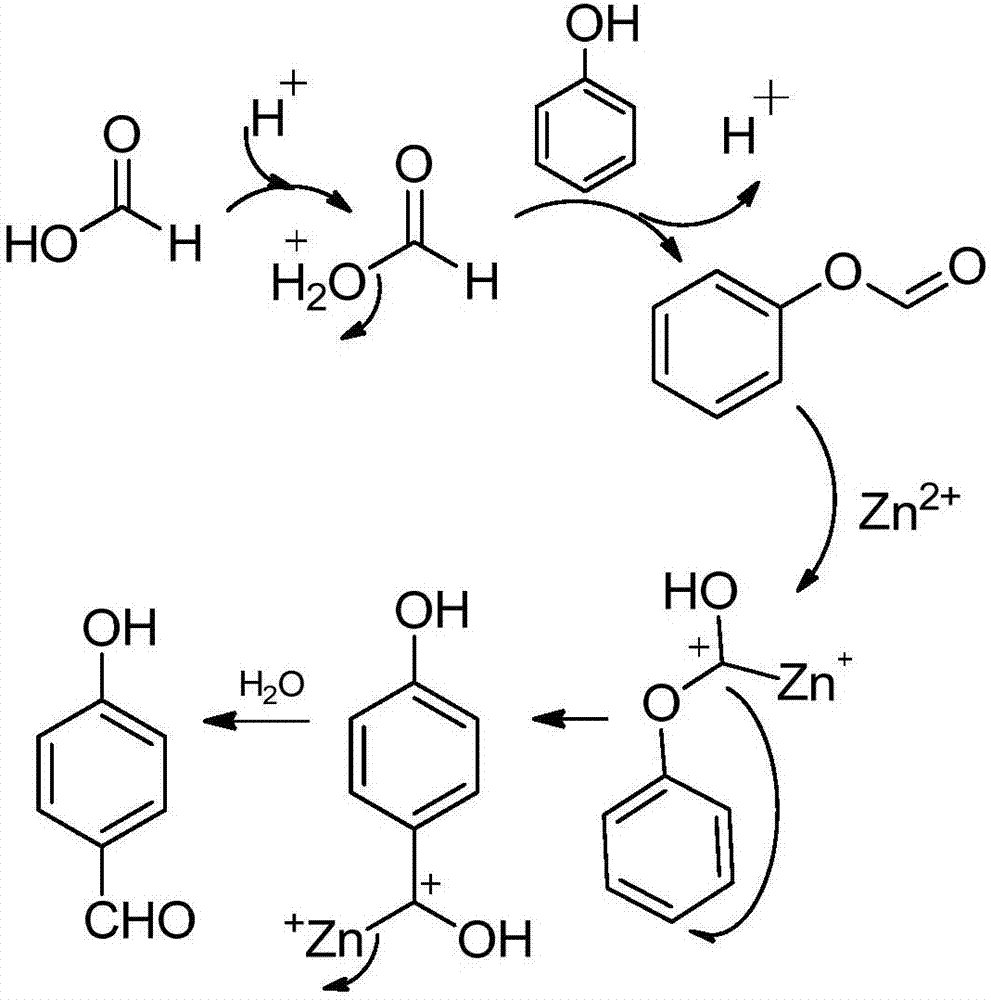
图2是本发明实施例1、3、4制备对羟基苯甲醛的反应机理图。
图3是本发明实施例2制备的对羟基苯甲醛的反应机理图。
具体实施方式
实施例1
如图1所示,本实施例的方法包括以下步骤:
步骤一、称取苯酚18.8g(0.2mol)、甲酸11.04g(0.24mol)、对甲苯磺酸3.8g(0.02mol)和甲苯200ml,加入到安装有加热套和冷凝回流器的反应瓶中,在磁力搅拌作用下,通过加热套将反应瓶中的反应液加热至105℃进行4h的酯化反应,酯化反应结束后停止加热,待反应液的温度降至40℃时,然后蒸馏反应液,收集171℃~173℃馏分,得到24.0g无色的甲酸苯酯,收率为98.4%,纯度为99.2%(gc纯度);
步骤二、称取甲酸苯酯12.2g(0.1mol)、氯化锌27.2g(0.2mol)和甲苯100ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至110℃进行4h的fries重排反应,fries重排反应结束后,停止加热并降温至40℃,然后向所述反应瓶中加入33ml去离子水,在磁力搅拌下加热,使体系温度升至60℃,保持搅拌30min,然后将加水后的反应液转移至分液漏斗中静置出现分层,然后收集有机相,最后对收集的有机相进行浓缩,除去有机相中的有机溶剂后得到浅黄色固体;
步骤三、用50ml混合溶剂对步骤二中得到的浅黄色固体进行重结晶,混合溶剂中乙醇和水的体积比1:1,得到11.8g白色的产物,产物经过气相色谱-质谱(gc-ms)表征:产物的分子量为122.0,与对羟基苯甲醛的理论分子量(122.12)相等,说明本实施例制备的产物确为对羟基苯甲醛;本实施例制备的对羟基苯甲醛的收率为96.7%,通过气相色谱法(gc)进行纯度测定为99.1%。
本实施例制备对羟基苯甲醛的反应机理为:如图2所示,首先是质子酸对甲酸进行活化,增强甲酸羰基的亲电反应性,活化的甲酸与苯酚的羟基发生酯化反应,得到甲酸苯基酯,同时释放出质子酸;甲酸苯基酯在锌离子的作用下,羰基与锌离子络合,同时锌离子的正电性传递给羰基碳原子,此时的羰基碳具有较强的亲电性,发生分子内的fries重排反应,选择性地重排到电子云密度较大的对位碳原子上,再释放出锌离子,得到对羟基苯甲醛。
实施例2
如图1所示,本实施例的方法包括以下步骤:
步骤一、称取苯酚18.8g(0.2mol)、甲酸11.04g(0.24mol)、浓硫酸2.0g(0.02mol,98%)和甲苯200ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至105℃;保持4h后,停止加热,使体系降温;体系温度降至40℃时,将体系蒸馏,收集171~173℃馏分,得到23.0g无色的甲酸苯酯,收率94.3%,纯度为99.1%(gc纯度);
步骤二、称取甲酸苯酯12.2g(0.1mol)、锌离子或三氯化铝26.7g(0.2mol)和甲苯100ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至110℃;保持4h后,停止加热,使体系降温;体系温度降至40℃时,给体系加入50ml质量分数10%的盐酸,磁力搅拌30min后,将体系转移至分液漏斗、静置,然后分出有机相;将所得有机相浓缩,除去溶剂,得到浅黄色固体;
步骤三、用乙醇和水的混合溶剂(体积比1:1)50ml对所得浅黄色固体进行重结晶,得到接近白色的产物11.5g,产物经过gc-ms表征:产物的分子量为122.0,与对羟基苯甲醛的理论分子量(122.12)相等,说明本实施例制备的产物确为对羟基苯甲醛;本实施例制备的对羟基苯甲醛的收率为94.3%,通过gc进行纯度测定为99.3%。
本实施例制备对羟基苯甲醛的反应机理为:如图3所示,首先是质子酸对甲酸进行活化,增强甲酸羰基的亲电反应性,活化的甲酸与苯酚的羟基发生酯化反应,得到甲酸苯基酯,同时释放出质子酸;甲酸苯基酯在三氯化铝的作用下,羰基与铝离子络合,同时铝离子的正电性传递给羰基碳原子,此时的羰基碳具有较强的亲电性,发生分子内的fries重排反应,选择性地重排到电子云密度较大的对位碳原子上,再释放出三氯化铝,得到对羟基苯甲醛。
实施例3
如图1所示,本实施例的方法包括以下步骤:
步骤一、称取苯酚18.8g(0.2mol)、甲酸11.04g(0.24mol)、对甲苯磺酸3.8g(0.02mol)和邻二氯苯200ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至180℃;保持4h后,停止加热,使体系降温;体系温度降至40℃时,将体系蒸馏,收集171~173℃馏分,得到24.0g无色甲酸苯酯液体,收率98.4%。
步骤二、称取甲酸苯酯12.2g(0.1mol)、氯化锌27.2g(0.2mol)和对二甲苯100ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至135℃;保持4h后,停止加热,使体系降温;体系温度降至40℃时,给体系加入66ml水,在磁力搅拌下给体系加热,使体系温度升至60℃;保持30min后,将体系转移至分液漏斗、静置,然后分出有机相;将所得有机相浓缩,除去溶剂,得到浅黄色固体;
步骤三、用乙醇和水的混合溶剂(体积比1:1)50ml对所得浅黄色固体进行重结晶,得到11.7g接近白色的产物,产物经过gc-ms表征:产物的分子量为122.0,与对羟基苯甲醛的理论分子量(122.12)相等,说明本实施例制备的产物确为对羟基苯甲醛;本实施例制备的对羟基苯甲醛的收率为95.9%,通过gc进行纯度测定为99.2%。
本实施例制备对羟基苯甲醛的反应机理与实施例1相同。
实施例4
如图1所示,本实施例的方法包括以下步骤:
步骤一、称取苯酚18.8g(0.2mol)、甲酸11.04g(0.24mol)、对甲苯磺酸3.8g(0.02mol)和对二甲苯200ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至138℃;保持4h后,停止加热,使体系降温;体系温度降至40℃时,将体系蒸馏,收集171~173℃馏分,得到24.0g无色甲酸苯酯液体,收率98.4%。
步骤二、称取甲酸苯酯12.2g(0.1mol)、氯化锌27.2g(0.2mol)和邻二氯苯100ml,加入到安装有加热套和冷凝回流器的反应瓶中,磁力搅拌下,加热至180℃;保持4h后,停止加热,使体系降温;体系温度降至40℃时,给体系加入100ml水,在磁力搅拌下给体系加热,使体系温度升至60℃;保持30min后,将体系转移至分液漏斗、静置,然后分出有机相;将所得有机相浓缩,除去溶剂,得到浅黄色固体;
步骤三、用乙醇和水的混合溶剂(体积比1:1)50ml对所得浅黄色固体进行重结晶,得到11.4g接近白色的产物,产物经过gc-ms表征:产物的分子量为122.0,与对羟基苯甲醛的理论分子量(122.12)相等,说明本实施例制备的产物确为对羟基苯甲醛;本实施例制备的对羟基苯甲醛的收率为93.4%,通过gc进行纯度测定为99.3%。
本实施例制备对羟基苯甲醛的反应机理与实施例1相同。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制。凡是根据发明技术实质对以上实施例所作的任何简单修改、变更以及等效变化,均仍属于本发明技术方案的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!