一种碳化硅陶瓷先驱体聚碳硅烷的制备方法与流程
本发明涉及碳化硅陶瓷领域,具体涉及一种碳化硅陶瓷先驱体聚碳硅烷的制备方法。
背景技术:
先驱体转化法制备陶瓷是20世纪70年代中期由德国verbeek和日本yajima等所创立的。先驱体转化高性能陶瓷材料可以灵活控制和改善陶瓷材料的化学结构、相组成、原子分布和微结构等,特别是在制备sic、si3n4和si-c-n等高性能非氧化物陶瓷方面具有传统陶瓷制备工艺所无法比拟的优势,受到学术界和工业界广泛关注。所谓先驱体转化法制备陶瓷是首先通过化学合成方法制得可经高温热解转化为陶瓷材料的聚合物,经成型后(包括纤维、粉体、涂层、多孔结构、以及复合材料等),再通过高温热解转化获得陶瓷材料。以先驱体转化法制备陶瓷材料,其关键之处在于能否制备出合适的先驱体,这直接决定了是否能成功制备出优异性能的陶瓷材料。
碳化硅(sic)陶瓷具有耐高温、抗氧化性优异、耐磨性好、热膨胀率小、热导率高、硬度高和耐腐蚀等优异特性,在航空航天、核电、高速刹车盘、热交换器等高端领域具有重要的应用价值。至今,有关sic陶瓷先驱体聚碳硅烷的结构和合成方法也层出不穷,归纳起来主要有以下几种途径:脱氯和热解重排、开环聚合、硅氢化加成反应、脱氢耦合、氯硅烷与炔化物缩聚、脱氯缩聚直接合成。
日本yajima利用二甲基二氯硅烷经碱金属脱氯缩合得到聚二甲基硅烷,然后经热解重排获得固态的聚碳硅烷(主要结构简式为[sihch3ch2]n)[chemicalletters,1975,9:931~934]。时至今日,这一结构的聚碳硅烷仍是最主要的sic陶瓷先驱体。但其仍存在一些不足,如:其结构中c/si为2,热解产物富碳,最终影响sic陶瓷的性能;陶瓷产率较低;在室温下为固体,用于制备复合材料或涂层等时,需要二甲苯、四氢呋喃等溶剂溶解,而在裂解之前又需要蒸发这些溶剂,导致制备周期长和工艺繁琐等。
开环聚合是利用环碳硅烷单体的开环反应制备聚碳硅烷,其原料多为杂环硅烷,在铂金等贵重金属催化剂作用下开环,形成si-c主链单体,进而聚合形成聚碳硅烷。如美国专利us4631179报道了采用1,3-二硅杂环丁烷在庚烷中用h2ptcl6催化开环聚合得到[sih2ch2]n结构的聚碳硅烷。liao等用正丁基锂引发四元或五元硅杂环发生阴离子开环聚合制得聚碳硅烷[macromolecules,1992,25:1639~1641;macromolecules,1992,25:3834~3837]。开环聚合工艺相对简单,但是其原料较难获得,导致制备成本较高,限制大规模生产和应用。
硅氢化加成反应是指含si-h化学键的化合物同不饱和有机基团在加热、光照或者催化剂作用下进行的加成反应,广泛应用于有机硅合成及改性。如boury等采用(ch2=ch)hsicl2在铂金属催化剂催化下进行自身硅氢加成反应,然后经还原得到结构为[sih2ch2ch2]n的聚碳硅烷[organometallics,1991,10:1457~1461]。pang等将乙炔基硅烷r2hsic≡ch在h2ptcl6催化下获得结构为[sirch3-ch=ch-]n的聚碳硅烷[macromolecules,1993,26:5671~5675]。但由于单体中c/si比高,易导致所得sic陶瓷中含有大量的游离碳。
zhang等采用二甲基二茂金属化合物催化含氢硅烷ch3sih3脱氢耦合制得结构为[sich3h]n的聚碳硅烷[journalofamericanceramicsociety,1991,74:670~673]。日本专利jp5209056a公开了一种由有机硅化合物h3si-a-sih3(a为亚甲基、2~7个c的亚甲基或烷基/三甲基硅基取代亚甲基、3~8个c的含二乙烯基的脂肪链)在催化剂(过渡金属化合物,比如hrh(co)(pph3)3、cp2zr(ch2sime3)2)作用下脱氢缩合得到聚碳硅烷的方法。脱氢耦合法副反应小产物纯度高,但含氢硅烷单体和催化剂易燃,制备难以操作。
在基于氯硅烷与炔化物缩聚路线制备聚碳硅烷应用方面,ijadi和corriu等用二氯硅烷与炔基双锂缩聚,得到结构式分别为[-sirr’-c≡c]n和[-(sirr’)m-c≡c-c≡c-]n的聚碳硅烷[journalofpolymersciencepartapolymerchemistry,1990,28:955~965;organometallics,1992,11:2507~2513]。但富碳的炔化物的引入也不易获得近化学计量比的sic陶瓷。
脱氯缩聚直接合成也是一种应用比较广泛的方法,如us5300614a公开了以氯甲基氯代硅烷(clch2sicl3)为原料,在醚类溶剂中依次经格式偶联反应和还原得到结构式为[h2sich2]n的聚碳硅烷。但由于si-cl键易与醚类溶剂反应,实际所得产物纯度并不高。cn104177621a主要以全部或部分烷氧基取代的氯甲基氯硅烷以及不饱和烷烃卤化镁为原料,通过格氏偶联反应和还原反应得到结构式为[sih2-nrnch2]m的聚碳硅烷(r独立地为含有c=c、c≡c或环丙基等的反应性基团,0.05≤n≤2)。该结构中同时含有si-h键和c=c等不饱和键,在一定条件下可自交联固化,具有较高的陶瓷产率。烷氧基的引入,虽然能抑制si-cl键与醚类溶剂反应,但也导致单体参与格式偶联反应活性低,不易引发。而且格式反应主要在醚类溶剂中进行,溶剂沸点普遍较低,即使引发后,由于格式反应放热剧烈,操作危险性高。
此外,日本专利jp6072704a通过阴离子聚合或者自由基聚合含乙烯基的硅烷化合物得到含有-sih2-ch2-ch2-或-sih2-ch(ch3)-等重复单元的聚碳硅烷。但由于重复单元中c/si为2,也不易获得近化学计量比的sic陶瓷。
综上所述,现有聚碳硅烷制备方法中,存在原料成本高、所得聚碳硅烷富碳或反应活性低等问题。
技术实现要素:
本发明的目的在于提供一种碳化硅陶瓷先驱体聚碳硅烷的制备方法,制备中所采用的原料来源简便、聚合反应活性高、以及可通过多种不同方式引入交联基团;制得的聚碳硅烷粘度低纯度高,经烧结后能得到近化学计量比的碳化硅陶瓷。
本发明提供如下技术方案:
一种碳化硅陶瓷先驱体聚碳硅烷的制备方法,包括以下步骤:
(1)将金属钠加入反应溶剂中,升温并逐步加入卤代甲基三烷氧基硅烷、或卤代甲基三烷氧基硅烷与三(卤代甲基)一烷氧基硅烷、或卤代甲基三烷氧基硅烷与四(卤代甲基)硅烷,反应生成物包括无机钠盐、含烷氧基的聚碳硅烷和烷氧基钠;
(2))去除步骤(1)中生成的无机钠盐和烷氧基钠,在环状醚类溶剂条件下,升温后加入还原试剂,含烷氧基的聚碳硅烷还原生成聚碳硅烷产物。
在步骤(1)中,升温至100~130℃,反应0.5~5小时。
在步骤(2)中,升温至40~80℃,反应3~10小时。
所述的卤素选自氯、溴或碘;所述的烷氧基选自甲氧基、乙氧基、丙氧基、丁氧基或异丙氧基。
所述的卤代甲基三烷氧基硅烷和三(卤代甲基)一烷氧基硅烷的结构分别为(ro)3si(ch2x)、(ro)si(ch2x)3,x为氯、溴或碘,r为甲基、乙基、丙基、丁基或异丙基。(ro)3si(ch2x)中含有卤代甲基硅结构(用≡si–ch2-x表示,x代表卤素)和烷氧基硅结构(用-si(or)3表示,or代表烷氧基),卤代甲基三烷氧基硅烷可简单通过卤代甲基三氯硅烷与醇在室温反应获得。
本发明提供的制备方法的反应原理为:
(ro)3si-ch2-x+2na→(ro)3si-ch2-na+nax(1)
(ro)3si-ch2-na+(ro)3si-ch2-na→(ro)3si-ch2-si(or)2-ch2-na+naor(2)
在步骤(1)中,(ro)3si(ch2x)与金属钠发生如式(1)所示反应,形成烷氧基硅取代的甲基钠((ro)3si-ch2-na),且脱除的卤素与金属钠形成盐沉淀,反应易引发。
烷氧基硅取代的甲基钠间发生如式(2)所示的反应:(ro)3si-ch2-na中同时含有-ch2-na结构和烷氧基硅结构,含有的-ch2-na具有非常强的亲核性,结构中的-ch2-na可与其它(ro)3si-ch2-na分子中的烷氧基硅反应,以及其结构中的烷氧基硅结构也可与其它(ro)3si-ch2-na分子中的-ch2-na结构发生亲核取代反应,从而形成烷氧基钠(用naor表示)和亚甲基连接的双烷氧基硅结构(即,主链主要为-si-ch2-si-ch2-si-ch2-结构且si连有or基团的大分子)。
通过步骤(2)中的还原反应,将与硅相连的or基团还原得到主链主要为-si-ch2-si-ch2-si-ch2-结构且si连有h基团的聚碳硅烷。所得聚碳硅烷中,c/si比接近1,这是获得具有近化学计量比sic陶瓷的保障。
优选的,在步骤(1)中,为避免反应过于剧烈,所述的卤代甲基三烷氧基硅烷中x为氯;为避免由于空间位阻导致后续还原不彻底和有利于后处理的顺利进行,所述的卤代甲基三烷氧基硅烷中的r为甲基或乙基。
优选的,在步骤(1)中同时加入三(卤代甲基)一烷氧基硅烷rosi(ch2x)3或四(卤代甲基)硅烷si(ch2x)4。超支化聚合物的球形结构使其不易发生链内缠结,内部存在大量的短支链结构,相对于相同分子量线性结构聚合物表现出更低的粘度,更好的流动性,所以可提高聚碳硅烷支化程度以改善其流动性和提高分子量。
所述的三(卤代甲基)一烷氧基硅烷rosi(ch2x)3的加入量为卤代甲基三烷氧基硅烷(ro)3sich2x摩尔量的1~10%。所述的四(卤代甲基)硅烷si(ch2x)4的加入量为卤代甲基三烷氧基硅烷(ro)3sich2x摩尔量的1~5%。
为避免提高c/si比,优选的,所述的三(卤代甲基)一烷氧基硅烷rosi(ch2x)3的加入量为卤代甲基三烷氧基硅烷(ro)3sich2x摩尔量的1~3%;所述的四(卤代甲基)硅烷si(ch2x)4的加入量为卤代甲基三烷氧基硅烷(ro)3sich2x摩尔量的1~2%。
优选的,在步骤(1)中,将金属钠和铜副族元素的混合物加入到反应溶剂中。铜副族元素的加入,有助于反应在低温引发,为利于金属钠的分散,反应后期在钠熔点之上反应。
在步骤(1)中,所述金属钠的加入量为卤代甲基三烷氧基硅烷、或卤代甲基三烷氧基硅烷与三(卤代甲基)一烷氧基硅烷、或卤代甲基三烷氧基硅烷与四(卤代甲基)硅烷中卤代甲基-ch2x摩尔量的2倍。
所述的铜副族元素为铜或银,加入量为金属钠摩尔量的1~5%。
优选的,为避免其它有机金属相关副反应的发生,所述的铜副族元素的加入量为金属钠摩尔量的1~3%。
优选的,在步骤(1)中,加入含不饱和基团的烷氧基硅烷、含不饱和基团的卤代烃或含不饱和基团的卤代烃的格式试剂。
所述的含不饱和基团的烷氧基硅烷选自乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、二乙烯基二甲氧基硅烷、二乙烯基二乙氧基硅烷、乙炔基三甲氧基硅烷、乙炔基三乙氧基硅烷、二乙炔基二甲氧基硅烷、二乙炔基二乙氧基硅烷、烯丙基三甲氧基硅烷、烯丙基三乙氧基硅烷、二烯丙基二甲氧基硅烷或二烯丙基二乙氧基硅烷中的一种或至少两种的组合。
所述的含不饱和基团的卤代烃选自3-氯丙烯、3-氯丙炔、溴代乙烯、3-溴丙烯或3-溴丙炔中的一种或至少两种的组合。所述的含不饱和基团的卤代烃的格式试剂选自乙烯基溴化镁、乙炔基氯化镁、乙炔基溴化镁、烯丙基氯化镁、烯丙基溴化镁、丙炔基氯化镁或丙炔基溴化镁中的一种或至少两种的组合。
所述的含不饱和基团的烷氧基硅烷、含不饱和基团的卤代烃或含不饱和基团的卤代烃的格式试剂的加入量为卤代甲基三烷氧基硅烷摩尔量的1/20~1/8。
优选的,所述的含不饱和基团的烷氧基硅烷、含不饱和基团的卤代烃或含不饱和基团的卤代烃的格式试剂的加入量为卤代甲基三烷氧基硅烷摩尔量的1/17~1/15,此投入量既能获得适宜的交联度又不明显增加所得碳化硅陶瓷中的化学计量比。
当采用添加含不饱和基团的卤代烃时,此时步骤(1)中还需要增加金属钠,增加的金属钠的加入量为含不饱和基团的卤代烃的摩尔量的2倍。
在步骤(2)中,去除烷氧基钠的分离方式为过滤或离心。
优选的,在步骤(2)中,去除烷氧基钠的方法为:在步骤(2)加入还原剂之前,加入三甲基氯硅烷,所述三甲基氯硅烷的摩尔量高于烷氧基钠。三甲基氯硅烷的沸点为57.3℃,将烷氧基钠转化成易沉淀分离的氯化钠,形成的三甲基烷氧基硅烷由于沸点低,也易通过蒸馏方式除去。
所述的制备方法包括对步骤(2)制备的聚碳硅烷产物进行后处理:通过蒸馏回收环状醚类溶剂,所得固体与盐酸水溶液混合,再加入萃取剂萃取水中的聚碳硅烷产物;将含聚碳硅烷产物的萃取剂用除水剂干燥,然后再经过蒸馏得到产物聚碳硅烷并回收萃取剂。
在步骤(1)中,所述的反应溶剂选自沸点高于金属钠的熔点,且在上述反应条件下与金属钠、卤代甲基三烷氧基硅烷等反应原料和反应中间产物不发生反应,以及具有较好溶解能力的反应溶剂,优选的,所述的反应溶剂选自正辛烷、异辛烷、正壬烷、异壬烷、2-甲基壬烷、甲苯、乙苯、二甲苯或环戊基甲醚中的一种或至少两种的组合。
在步骤(2)中先去除反应溶剂,所述的环状醚类溶剂选自四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环或环戊基甲醚中的一种或至少两种的组合。
在步骤(2)中,所述的还原试剂选自氢化锂、氢化铝锂或氢化钠。优选的,所述的还原试剂为氢化铝锂,其用量为还原步骤(1)后剩余烷氧基硅结构所需理论量的1.2~1.5倍。优选的,为减少后处理中盐酸的消耗,氢化铝锂用量为还原步骤(1)后剩余烷氧基硅结构所需理论量的1.2~1.3倍。
在后处理中,所述的萃取剂选自对反应产物有较好溶解能力且疏水的萃取剂,优选为乙醚、石油醚、戊烷、正己烷、环己烷、正庚烷或环戊基甲醚中的一种或至少两种的组合。
进一步优选的,所述反应溶剂、环状醚类溶剂和萃取剂均为环戊基甲醚。环戊基甲醚是一种环保高性能疏水性醚类溶剂(密度:0.86g/cm3;沸点:106℃),作为反应溶剂,可用于格式反应、偶联反应、偶联胺化反应、金属还原反应、路易斯酸反应和弗里德-克拉夫茨反应等多种反应;同时也可用于萃取、结晶、表面处理和聚合过程。当采用环戊基甲醚为溶剂时,在步骤(2)中,无需对反应溶剂进行蒸馏回收,去除钠盐后直接进行还原。
在后处理中,所述的干燥剂选自硫酸钠、硫酸镁、氯化钙、硫酸钙或分子筛中的一种或至少两种的组合。优选的,所述的干燥剂为硫酸镁,其吸水速率快和吸附容量较大。
同现有技术相比,本发明的有益效果体现在:
本发明以廉价易得的卤代甲基烷氧基硅烷为主要原料,经形成相应的烷基钠后,可相互发生亲核取代反应,再经还原后可获得c/si小于1.4的聚碳硅烷,烧结后生成的碳化硅陶瓷为近化学计量比。与金属镁相比,钠活性更高,而且形成的烷基钠试剂也有很高的亲核性,所以整个聚合反应活性高易进行。通过对溶剂的优化、去除烷氧基钠和引入不饱和基团方式的优化,使工艺得到简化。此外,所采用的原料来源简便、制得的聚碳硅烷粘度低纯度高,经烧结后能得到近化学计量比的碳化硅陶瓷。
附图说明
图1为实施例1所得聚碳硅烷的核磁氢谱;
图2为实施例1所得聚碳硅烷的红外光谱;
图3为实施例2所得聚碳硅烷的核磁氢谱;
图4为实施例2所得聚碳硅烷的红外光谱;
图5为实施例2所得聚碳硅烷经1600℃烧结后所得陶瓷xrd曲线;
图6为实施例4所得聚碳硅烷的核磁氢谱;
图7为实施例4所得聚碳硅烷的红外光谱,(a)为未经交联;(b)为交联后。
具体实施方式
以下结合具体实施例对本发明作进一步详细描述,有必要指出的是本实施例只用于对本发明进行进一步的说明,不能理解为对本发明保护范围的限制。
对本发明制备的聚碳硅烷进行如下性能测试:
红外光谱分析:采用美国thermonicolet6700傅立叶变换红外光谱分析仪测试;
核磁氢谱分析:采用德国bruker公司400mhzavanceⅲ型核磁共振谱仪测试;
元素含量分析:碳元素采用美国leco公司cs844型红外吸收碳硫分析仪测试、氧元素采用日本horiba公司emga-620型氧氮分析仪测试、h元素采用美国perkinelmer公司pe2400ⅱ型chns/o有机元素分析仪测试;
数均分子量:采用日本tosoh公司hlc-8320gpc型凝胶渗透色谱仪测试;
粘度:采用德国thermoscientiftc公司marsiii型流变仪测试;
x-射线衍射(xrd):采用德国bruker公司d8advance型x射线衍射仪测试;
陶瓷产率:采用日本精工生产tg-dta6300型热重分析仪(tga)测试。
实施例1
结构式为[sih2ch2]n的聚碳硅烷的制备。
在氮气保护和室温环境下,向带机械搅拌器、温度计、氮气导入管、冷凝器和滴液漏斗的四口烧瓶中,将金属钠(1mol)和二甲苯(500ml)加入,搅拌并升温至100℃。待温度恒定后,将氯代甲基三甲氧基硅烷(0.5mol)分批加入,之后继续反应3小时。待溶液温度降至室温后,将上层溶液通过倾倒转移至离心管中,再高速离心除去其中残留的固态悬浮物。通过减压蒸馏将溶剂二甲苯回收,向所得中间产物中加入经钠除水的四氢呋喃(500ml),升温至40℃,然后分批加入还原试剂氢化铝锂(0.35mol),再继续于60℃反应5小时。再通过旋转蒸馏将四氢呋喃回收,所得固体分批加入盐酸冰水溶液中(浓度为4mol/l,体积为200ml)并加入正己烷(300ml),然后通过分液漏斗除去水相,有机相再经过3次水洗、硫酸钠干燥和蒸馏得到液态超支化聚碳硅烷。
对制备的聚碳硅烷进行如下性能测试:
数均分子量为752g/mol;依据元素含量分析得出分子式为sic0.98h3.89o0.005;粘度:12mpa·s。逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为52%,根据元素组成得出其分子式为sic1.0o0.03。
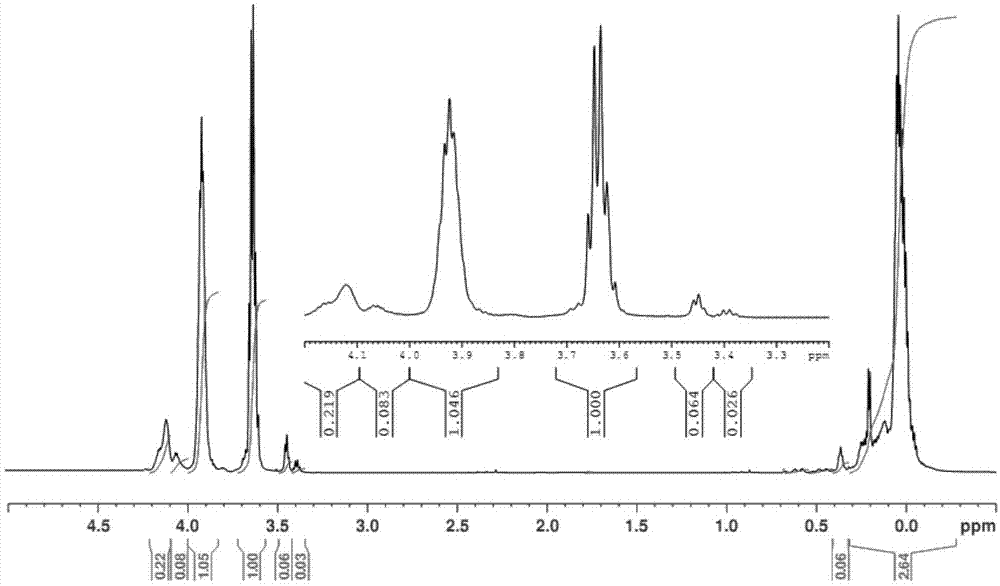
图1为所得聚碳硅烷的核磁氢谱,具体信号归属为:3.6ppm为si-h3信号峰,3.9ppm为si-h2信号峰,4.1ppm为si-h信号峰,0ppm附近为si-ch2-信号峰。
图2为所得聚碳硅烷的红外光谱,具体信号归属为:842cm-1(si-cstretching),933cm-1(si-h2stretching),1048cm-1(si-ch2-sistretching),1357cm-1(si-ch2-sideformation),2132cm-1(si-hstretching),2922cm-1和2876cm-1(c-hstretchinginsi-ch2)。
实施例2
结构式为{[sih2ch2]9-[sih(ch2-ch=ch2)ch2]1}n的聚碳硅烷的制备。
在氮气保护和室温环境下,向带机械搅拌器、温度计、氮气导入管、冷凝器和滴液漏斗的四口烧瓶中,将金属钠(1mol)、氯丙烯(0.05mol)、尺寸为10~30nm的铜粉(0.01mol)、氯代甲基三甲氧基硅烷(0.05mol)和环戊基甲醚(500ml)加入,搅拌并升温至100℃。待温度恒定后,将氯代甲基三甲氧基硅烷(0.4mol)分批加入,之后继续反应4小时。待溶液温度降至室温后,加入三甲基氯硅烷(0.6mol)并搅拌0.5小时,然后在35℃通过循环水泵(真空度0.095mpa)抽出三甲基氯硅烷和三甲基一甲氧基硅烷。将所得溶液升温至50℃,然后分批加入还原试剂氢化铝锂(0.26mol),再继续于60℃反应5小时。冷却后,分批加入盐酸冰水溶液中(浓度为4mol/l,体积为200ml),然后通过分液漏斗除去水相,有机相再经过3次水洗、硫酸镁干燥和蒸馏得到含烯丙基的液态聚碳硅烷。
对制备的聚碳硅烷进行如下性能测试:
数均分子量为886g/mol;依据元素含量分析得出分子式为sic1.21h4.25o0.003;粘度:25mpa·s。
附图3为所得聚碳硅烷的核磁氢谱,具体信号归属为:3.6ppm为si-h3信号峰,3.9ppm为si-h2信号峰,4.1ppm为si-h信号峰,0ppm附近为si-ch2-信号峰,1.7ppm、4.9ppm和5.8ppm分别为烯丙基结构中的-ch2-、-c=ch2和c=ch-信号峰。附图4为所得聚碳硅烷的红外光谱,具体信号归属为:842cm-1(si-cstretching),934cm-1(si-h2stretching),1048cm-1(si-ch2-sistretching),1357cm-1(si-ch2-sideformation),1630cm-1(c=cstretchinginsi-ch2-ch=ch2),2130cm-1(si-hstretching),2920cm-1和2872cm-1(c-hstretchinginsi-ch2),3080cm-1(=c-hstretchinginsi-ch2-ch=ch2)。
取5.0g所得含烯丙基的液态聚碳硅烷,加入质量为0.05g的过氧化苯甲酸叔丁酯,混合均匀,然后于80℃保温1小时,得到交联固化的聚碳硅烷。然后逐步升温至1600℃,得到黑色的陶瓷产物,陶瓷产率为72%。根据元素组成得出其分子式为sic1.08o0.04,附图5为其xrd曲线,证实为sic陶瓷。
实施例3
结构式为{[sih2ch2]9-[sih(ch2-ch=ch2)ch2]1}n的超支化聚碳硅烷的制备:
在氮气保护和室温环境下,向带机械搅拌器、温度计、氮气导入管、冷凝器和滴液漏斗的四口烧瓶中,将金属钠(1mol)、氯丙烯(0.05mol)、尺寸为10~30nm的铜粉(0.05mol)和环戊基甲醚(500ml)加入,搅拌并升温至100℃。待温度恒定后,将氯代甲基三甲氧基硅烷(0.42mol)和三(氯代甲基)一甲氧基硅烷(0.01mol)的混合物分批加入,之后继续反应3小时。待溶液温度降至室温后,加入三甲基氯硅烷(0.6mol)并搅拌0.5小时,然后在35℃通过循环水泵(真空度0.095mpa)抽出三甲基氯硅烷和三甲基一甲氧基硅烷。将所得溶液升温至50℃,然后分批加入还原试剂氢化铝锂(0.26mol),再继续于60℃反应5小时。冷却后,分批加入盐酸冰水溶液中(浓度为4mol/l,体积为200ml),然后通过分液漏斗除去水相,有机相再经过3次水洗、硫酸钠干燥和蒸馏得到含烯丙基的液态聚碳硅烷。
检测结果:
数均分子量为1326g/mol;依据元素含量分析得出分子式为sic1.27h4.42o0.003;粘度:24mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为76%,根据元素组成得出其分子式为sic1.10o0.03。
实施例4
结构式为{[sih2ch2]9-[sih(c≡ch)ch2]1}n的聚碳硅烷的制备。
在氮气保护和室温环境下,向带机械搅拌器、温度计、氮气导入管、冷凝器和滴液漏斗的四口烧瓶中,将金属钠(1mol)、尺寸为10~30nm的铜粉(0.03mol)、氯代甲基三乙氧基硅烷(0.05mol)和环戊基甲醚(500ml)加入,搅拌并升温至100℃。待温度恒定后,将氯代甲基三乙氧基硅烷(0.45mol)分批加入,之后继续反应3小时。待溶液温度降至室温后,加入三甲基氯硅烷(0.6mol)并搅拌0.5小时,然后在35℃通过循环水泵(真空度0.095mpa)抽出三甲基氯硅烷和三甲基一甲氧基硅烷。之后加入乙炔基溴化镁的环戊基甲醚溶液(浓度为0.5mol/l,加入量为60ml),并于60℃保温1小时。将所得溶液降温至50℃,然后分批加入还原试剂氢化铝锂(0.29mol),再继续于60℃反应5小时。冷却后,分批加入盐酸冰水溶液中(浓度为4mol/l,体积为200ml),然后通过分液漏斗除去水相,有机相再经过3次水洗、硫酸钠干燥和蒸馏得到含乙炔基的液态聚碳硅烷。
检测结果:
数均分子量为1024g/mol;依据元素含量分析得出分子式为sic1.08h4.03o0.004;粘度:32mpa·s;
图6为所得聚碳硅烷的核磁氢谱,具体信号归属为:3.6ppm为si-h3信号峰,3.9ppm为si-h2信号峰,4.1ppm为si-h信号峰,2.5ppm为c≡ch信号峰,0ppm附近为si-ch2-信号峰。图7(a)为所得聚碳硅烷的红外光谱,具体信号归属为:842cm-1(si-cstretching),933cm-1(si-h2stretching),1048cm-1(si-ch2-sistretching),1357cm-1(si-ch2-sideformation),2037cm-1(c≡cstretching),2140cm-1(si-hstretching),2922cm-1和2881cm-1(c-hstretchinginsi-ch2),3288cm-1(-c-hstretchingin-c≡c-h)。
取5.0g所得含乙炔基的液态聚碳硅烷,加入质量为0.05g的过氧化苯甲酸叔丁酯,混合均匀,然后于120℃保温1小时,得到交联固化的聚碳硅烷,从图7(b)可看出,交联后乙炔基信号峰消失。然后逐步升温至1600℃,得到黑色的陶瓷产物,陶瓷产率为74%。根据元素组成得出其分子式为sic1.01o0.03,xrd证实为sic陶瓷。
实施例5
如实施例3提供的制备方法,加入氯丙烯(0.0185mol)、尺寸为10~30nm的银粉(0.01mol)、氯代甲基三甲氧基硅烷0.37mol和三(氯代甲基)一甲氧基硅烷0.037mol。还原时,氢化铝锂加入量为0.195mol。
所得聚碳硅烷数均分子量为1671g/mol;依据元素含量分析得出分子式为sic1.30h4.47o0.003;粘度:27mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为71%,根据元素组成得出其分子式为sic1.13o0.05。
实施例6
如实施例2提供的制备方法,加入氯丙烯(0.03mol)、尺寸为10~30nm的银粉(0.01mol)、氯代甲基三甲氧基硅烷0.45mol和三(氯代甲基)一甲氧基硅烷0.007mol。还原时,氢化铝锂加入量为0.23mol。
所得聚碳硅烷数均分子量为931g/mol;依据元素含量分析得出分子式为sic1.22h4.30o0.004;粘度:14mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为78%,根据元素组成得出其分子式为sic1.07o0.03。
实施例7
如实施例3提供的制备方法,加入尺寸为10~30nm的银粉(0.03mol)、氯代甲基三甲氧基硅烷0.405mol和三(氯代甲基)一甲氧基硅烷0.015mol。还原时,氢化铝锂加入量为0.22mol。
检测结果:
数均分子量为1139g/mol;依据元素含量分析得出分子式为sic1.36h4.53o0.003;粘度:23mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为70%,根据元素组成得出其分子式为sic1.17o0.03。
实施例8
如实施例3提供的制备方法,加入氯丙烯(0.03mol)、尺寸为10~30nm的铜粉(0.01mol)、氯代甲基三甲氧基硅烷0.456mol和三(氯代甲基)一甲氧基硅烷0.0046mol。还原时,氢化铝锂加入量为0.26mol。
检测结果:
数均分子量为958g/mol;依据元素含量分析得出分子式为sic1.18h4.19o0.003;粘度:19mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为77%,根据元素组成得出其分子式为sic1.04o0.03。
实施例9
如实施例3提供的制备方法,加入氯丙烯(0.04mol)、尺寸为10~30nm的铜粉(0.01mol)、氯代甲基三甲氧基硅烷0.42mol和si(ch2cl)40.01mol。还原时,氢化铝锂加入量为0.24mol。
检测结果:
所得数均分子量为1006g/mol;依据元素含量分析得出分子式为sic1.35h4.44o0.005;粘度:18mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为69%,根据元素组成得出其分子式为sic1.15o0.03。
实施例10
如实施例3提供的制备方法,加入乙烯基三甲氧基硅烷(0.03mol)替代氯丙烯、尺寸为10~30nm的银粉(0.01mol)、氯代甲基三甲氧基硅烷0.45mol和si(ch2cl)40.005mol。还原时,氢化铝锂加入量为0.29mol。检测结果:
所得数均分子量为1957g/mol;依据元素含量分析得出分子式为sic1.08h4.05o0.003;粘度:38mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为77%,根据元素组成得出其分子式为sic1.03o0.05。
实施例11
如实施例3提供的制备方法,加入乙烯基三甲氧基硅烷(0.026mol)替代氯丙烯、尺寸为10~30nm的银粉(0.01mol)、氯代甲基三甲氧基硅烷0.44mol和si(ch2cl)40.0085mol。
检测结果:还原时,氢化铝锂加入量为0.25mol。
所得数均分子量为2318g/mol;依据元素含量分析得出分子式为sic1.11h4.15o0.002;粘度:37mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为76%,根据元素组成得出其分子式为sic1.04o0.04。
实施例12
如实施例3提供的制备方法,加入氯丙烯(0.026mol)、尺寸为10~30nm的铜粉(0.02mol)、氯代甲基三甲氧基硅烷0.435mol和三(氯代甲基)一甲氧基硅烷0.013mol。还原时,氢化铝锂加入量为0.24mol。
检测结果:
数均分子量为1235g/mol;依据元素含量分析得出分子式为sic1.19h4.17o0.004;粘度:20mpa·s。加入1wt%的过氧化苯甲酸叔丁酯并逐步升温至1600℃,得到黑色的碳化硅,陶瓷产率为78%,根据元素组成得出其分子式为sic1.06o0.03。
本发明制备的聚碳硅烷烧结后的碳化硅陶瓷化产率较高,同时烧结后的碳化硅陶瓷的化学计量比为1:1-1.17。特别的,在制备方法中采用优选方案,制备的聚碳硅烷烧结后的化学计量比为1:01-1.07,更接近1::1,陶瓷化产率为76-78%。
- 还没有人留言评论。精彩留言会获得点赞!