一种氟碳链改性亚磷酸酯类丙烯酸酯化合物及其制备方法和应用与流程
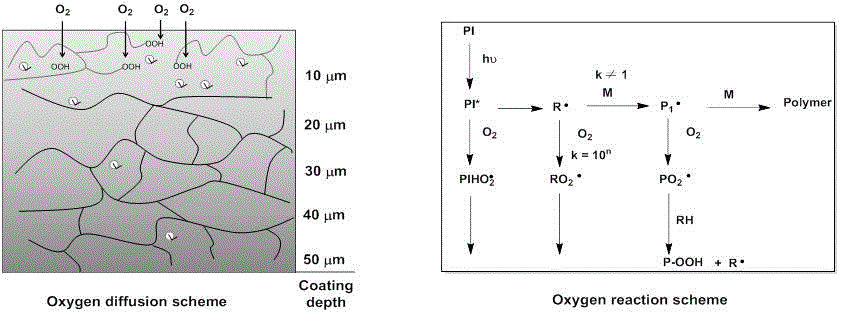
本发明涉及高分子材料光固化领域,更具体地,涉及一种氟碳链改性亚磷酸酯类丙烯酸酯化合物及其制备方法和应用。技术背景自由基聚合是光固化技术当中应用得最为广发的技术之一。传统自由基型光引发剂引发聚合反应,尽管固化速度快,但是自由基聚合反应普遍存在的氧阻聚效应,从而光固化涂层表面始终存在不容忽视的氧阻聚,导致涂层表面聚合交联不充分,出现表层耐磨性不足,甚至表面发粘、返粘等弊病。有效抑制或减少表面氧阻聚可有效解决由于表面固化不完全导致材料表面性能变差的问题。能有效抑制或减少表面氧阻聚的办法是:1)峰值照射,即采用价格低廉、节能、放热少以及光强高的395nm和405nm的uv-led进行光固化反应;2)加入助剂添加剂,氢供体(比如胺类,巯基类,硅烷类等氢供体)化合物,比如叔胺类,能有效的与空气中存在的三线态氧气分子反应,有效抑制氧阻聚,同时能加快typeⅱ型光引发剂分解速率,加速完成反应(如图1);或还原剂(如亚磷酸酯类)类助剂,能有效与过氧化物反生氧化还原反应,释放活性自由基,继续引发聚合(亚磷酸酯类助剂抗氧阻聚的作用机理,如图2)。方案1氧阻聚的机理解释以及迁移氧气策略:(a)光引发剂激发态的猝灭,(b)引发或增长自由基形成惰性的过氧自由基,(c)开始阶段的氧阻聚解决方案(如惰性环境、光源、分子内形成的惰性环境以及光引发剂等),(d)单重态氧气清除剂,(e)还原剂,(f)氢供体,(g)自由基-自由基耦合终止,(h)氢提取,(i)过氧化物分解,(j)清除氧气分子,(k)再次进行聚合。虽然使用叔胺类助剂能有效抑制氧阻聚,但其会使材料发生黄变,影响材料尤其是透明无色材料的美观,达不到性能要求;硫醇类助剂虽然能抑制氧阻聚,但是其加工稳定性很差。且上述两种助剂通常有很强的刺激性气味。亚磷酸的氧化产物磷酸酯p=o键极为稳定,即亚磷酸酯类助剂具有较强的氧化还原作用,并且其氧化产物无色,即亚磷酸酯类助剂具有耐变色和无气味性等自身优异特性,达到抑制氧阻聚的同时不影响材料的色泽,提高材料的耐热稳定性;亚磷酸酯类助剂可通过还原聚合物类过氧化物,使聚合物变成稳定的醇类聚合物,从而提高聚合物的耐候性。亚磷酸酯类助剂抑制氧阻聚的作用机理如下:亚磷酸酯类助剂可以与氧气直接结合,消耗氧气。或者与过氧自由基、烷氧自由基发生氧化还原反应,将活性自由基重新释放,再次引发聚合反应。氟碳长链化合物具有降低材料表面张力,提高材料的疏水疏油能力,从而提高材料表层的耐污能力,而广泛应用各种耐污材料,疏水材料中。故为了有效抑制或克服氧阻聚,以及解决涂层固化时的黄变问题,从而提高涂层表观性能,有必要研发出新型的具有低表面张力的耐黄变丙烯酸酯单体,应对表面氧阻聚问题。技术实现要素:本发明的目的在于克服现有技术中丙烯酸酯单体存在氧阻聚,及涂层固化时出现黄变的缺陷或不足,提供一种氟碳链改性亚磷酸酯类丙烯酸酯化合物。本发明提供的氟碳链改性亚磷酸酯类丙烯酸酯化合物含有亚磷酸酯基中心和全氟烷基链,在体系中可快速上浮到涂层表面,同时亚磷酸酯基可与氧气发生氧化还原作用达到抑制表面氧阻聚的作用,使表面固化完善,不黄变,使表观性能良好;全氟烷基链介入交联网络的概率大,牢固度提高,可赋予材料防污耐擦拭性;同时亚磷酸酯类化合物可以通过还原作用,将聚合物变成稳定的醇类聚合物,从而提高材料的耐候性。本发明的另一目的在于提供上述氟碳链改性亚磷酸酯类丙烯酸酯化合物的制备方法。本发明的另一目的在于提供上述氟碳链改性亚磷酸酯类丙烯酸酯化合物在制备光固化材料中的应用。为了实现上述发明目的,本发明采用如下技术方案:一种氟碳链改性亚磷酸酯类丙烯酸酯化合物,具有如式(ⅰ)所示结构:其中,r2包含(甲基)丙烯酸酯类双键结构;r3为a为c1~5的直链或支链烷氧基,n为1~18;r1为-o-r2、-o-r3、c1~8的直链或支链烷基、苯基、取代苯基或c1~8的直链或支链烷基。本发明提供的氟碳链改性亚磷酸酯类丙烯酸酯化合物通过接入具有低表面张力的全氟烷基链,使得该化合物在配方体系中得以上浮至涂层/油墨表面,其亚磷酸酯基结构使得其可以与过氧化物发生氧化还原反应,重新释放出活性自由基,继续引发聚合,抑制氧阻聚;同时,具有亚磷酸酯基结构的丙烯酸酯类单体不发生黄变,不影响透明或浅色体系材料的表观性能。亚磷酸酯基的引入有效抑制表面氧阻聚,使得全氟烷基链接入交联网络概率增大,牢固度提高,防污耐擦拭性能可显著提高,提高材料耐候性。优选地,r2为(甲基)丙烯酸乙酯基、(甲基)丙烯酸丙酯基、2-甲基-2-丙烯酸-1,2-丙二醇酯、2-丙烯酸-2-羟基-1,3-丙二酯或2,3-二羟基丙烷丙烯酸酯。优选地,n为12~18;a为丙基、乙基、丙基、异丙基或丁基。优选地,r1为乙基、丙基、丁基、苯基、异戊基、4-甲氧基苯基、环己基、苄基、-o-r2或-o-r3。具体地,氟碳链改性亚磷酸酯类丙烯酸酯化合物为c2~8的脂肪族丙烯酸羟酯类化合物、c2~6的羟基脂肪族二醇二(甲基)丙烯酸酯、c3~6的含羟基的脂肪族三元醇三丙烯酸酯、c3~6的羟基二(甲基)丙烯酸酯、羟基多缩乙二醇与羟基多缩丙二醇等低聚醚二元醇的丙烯酸酯。上述氟碳链改性亚磷酸酯类丙烯酸酯化合物的制备方法,包括如下步骤:式(ⅱ)所示的二卤化磷化合物与含羟基的丙烯酸酯化合物在缚酸剂存在的条件下,发生取代反应生成如式(ⅲ)所示的化合物;然后加入如式(ⅳ)所示的全氟烷基醇,发生取代反应,即得所述氟碳链改性亚磷酸酯类丙烯酸酯化合物;x为卤素,r为x或c1~8的直链或支链烷基、苯基、取代苯基或c1~8的直链或支链烷基。本发明s3步骤中既可以先滴加含羟基的丙烯酸酯化合物至二卤化磷化合物中,又可以先滴加全氟烷基醇至二卤化磷化合物中。三者先发生取代反应,生成式(ⅲ)的化合物,再进一步取代得到氟碳链改性亚磷酸酯类丙烯酸酯化合物。优选地,x为溴或氯。优选地,所述二卤化磷化合物为三氯化磷、1,4-双(二氯磷基)苯、二氯丙基磷化氢、1-(二氯磷基)-4-甲氧基苯基、环己二氯磷、(三氟甲基)二氯化磷、苄基二氯化磷、异戊基二氯化磷、乙基二氯化磷、三溴化磷、乙基二溴化磷、二溴(全氟苯基)化磷或二溴苯基。具体地,二卤化磷化合物的结构式如下:优选地,所述含羟基的丙烯酸酯化合物为丙烯酸羟乙酯、甲基丙烯酸羟丙酯、甲基丙烯酸羟乙酯、2-甲基-2-丙烯酸-1,2-丙二醇酯、2-丙烯酸-2-羟基-1,3-丙二酯、2,3-二羟基丙烷丙烯酸酯或季戊四醇三丙烯酸酯。具体地,含羟基的丙烯酸酯化合物的结构式如下:优选地,所述缚酸剂为叔胺类。更为优选地,所述叔胺类为三乙胺、n,n-二甲基苄胺或n,n-二甲基正丁胺中的一种或几种。优选地,所述取代反应选用甲苯、四氢呋喃、乙腈、乙醚或氯仿中的一种或几种作为溶剂。优选地,s1中在-10~10℃下将含羟基的丙烯酸酯化合物滴加至二卤化磷化合物中;s3中在-10~10℃下将含羟基的丙烯酸酯化合物、全氟烷基醇滴加至二卤化磷化合物中。优选地,所述二卤化磷化合物和含羟基的丙烯酸酯化合物的摩尔比为1.0:1.0~2.0;所述二卤化磷化合物和全氟烷基醇的摩尔比为1.0:1.0。优选地,s1~s3中所述取代反应的温度为30~120℃,反应的时间为3~5h;所述取代反应在氮气气氛下进行。上述氟碳链改性亚磷酸酯类丙烯酸酯化合物在制备光固化材料中的应用也在本发明的保护范围内。与现有技术相比,本发明具有如下有益效果:本发明提供的氟碳链改性亚磷酸酯类丙烯酸酯化合物通过接入具有低表面张力的聚硅氧烷链,使得单体在配方体系中得以上浮至涂层/油墨表面,其亚磷酸酯基结构使得其可以与过氧化物发生氧化还原反应,重新释放出活性自由基,继续引发聚合,抑制氧阻聚;同时,具有亚磷酸酯基结构的化合物与氧气作用后生成的物质为磷酸酯类化合物,该化合物无色,即使用该助剂不发生黄变,不影响透明或浅色体系材料的表观性能。亚磷酸酯基的引入有效抑制表面氧阻聚,使得全氟烷基链接入交联网络概率增大,牢固度提高,防污耐擦拭性能可显著提高。具体实施方式下面结合实施例进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明的范围。下例实施例中未注明具体条件的实验方法,通常按照本领域常规条件或按照制造厂商建议的条件;所使用的原料、试剂等,如无特殊说明,均为可从常规市场等商业途径得到的原料和试剂。本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。附图说明图1为氧阻聚机理;图2为亚磷酸酯类助剂抗氧阻聚的作用机理。实施例1本实施例提供一种上述氟碳链改性亚磷酸酯类丙烯酸酯化合物,为亚磷酸酯丙烯酸酯类单体,其结构式如下:该单体通过如下方法制备得到。取带有回流、滴加、搅拌的标准反应装置,置于-10℃的环境中。将三氯化磷(10g,0.0728mol)的50ml无水乙醚溶液,三乙胺(0.220mol,23g)加入到三口烧瓶中,氮气氛围下,当温度稳定在-10℃时,在机械搅拌下再逐滴滴加全氟辛基乙醇(0.073mol,34g),滴加过程控制温度-10℃-0℃。滴加完成后,此温度下继续搅拌30min。再逐滴滴加丙烯酸羟乙酯hea(0.15mol,17.4g),滴加过程控制温度-10℃-0℃。滴加完成后,缓慢升温到60℃左右,保温反应3h。反应结束后冷却,过滤除去三乙胺盐酸盐,滤液减压蒸除溶剂和过量三乙胺,得化合物m1,稍微过量的hea可以作为活性稀释剂在体系中共同使用。核磁数据表征为1hnmr(cdcl3,400mhz):δ6.42(d,2h),6.15(q,2h),5.83(d,2h),4,29(t,4h),3.79(m,6h),1.83(m,2h).核磁表明成功合成了对应化合物m1。实施例2本实施例提供一种述氟碳链改性亚磷酸酯类丙烯酸酯化合物,为亚磷酸酯丙烯酸酯类单体,其结构式如下:该单体通过如下方法制备得到。取带有回流、滴加、搅拌的标准反应装置,置于-5℃的冷水浴中。将二苯基氯化磷(0.03mol,10g)的50ml甲苯溶液,吡啶(0.07mol,5.6g)加入到三口烧瓶中,氮气氛围下,当温度稳定在-5℃时,在机械搅拌下逐滴滴加全氟辛基丙醇(0.028mol,13.5g),滴加完成后,此温度下继续搅拌30min。再逐滴滴加丙烯酸羟乙酯hea(0.035mol,5.8g),滴加过程控制温度-5℃-0℃。完全滴加完成后,缓慢升温到80℃左右,保温反应3h。反应结束后冷却,过滤除去吡啶盐酸盐,滤液减压蒸除溶剂和过量吡啶,得化合物m1,稍微过量的hea可以作为活性稀释剂在体系中共同使用。核磁数据表征为1hnmr(cdcl3,400mhz):δ7.43(m,3h),7.15(t,2h)6.41(d,1h),6.12(q,1h),5.82(d,1h),4.30(t,2h),3.79(q,2h),1.58(m,2h),1.50(m,2h).核磁表明成功合成了对应化合物m2。实施例3本实施例提供一种述氟碳链改性亚磷酸酯类丙烯酸酯化合物,为亚磷酸酯丙烯酸酯类单体,其结构式如下:该单体通过如下方法制备得到。取带有回流、滴加、搅拌的标准反应装置,置于-5℃的冷水浴中。将乙基二溴化磷(0.03mol,6.57g)的50ml乙腈溶液,三乙胺(0.07mol,7g)加入到三口烧瓶中,氮气氛围下,当温度稳定在-5℃时,在机械搅拌下逐滴滴加全氟癸基乙醇(0.028mol,15.5g),滴加完成后,此温度下继续搅拌30min。再逐滴滴加2-丙烯酸-2-羟基-1,3-丙二酯(0.035mol,8g),滴加过程控制温度-5℃-0℃。完全滴加完成后,缓慢升温到75℃左右,保温反应3h。反应结束后冷却,过滤除去三乙胺盐酸盐,滤液减压蒸除溶剂和过量三乙胺,得化合物m3,稍微过量的2-丙烯酸-2-羟基-1,3-丙二酯可以作为活性稀释剂在体系中共同使用。核磁数据表征为1hnmr(cdcl3,400mhz):δ6.42(d,2h),6.13(q,2h),5.84(d,2h),4.83(m,1h),4.29(d,4h)3.83(q,2h),2.13(m,2h),1.58(m,2h),1.53(m,2h),0.98(t,3h).核磁表明成功合成了对应化合物m3。实施例4本实施例提供一种述氟碳链改性亚磷酸酯类丙烯酸酯化合物,为亚磷酸酯丙烯酸酯类单体,其结构式如下:该单体通过如下方法制备得到。取带有回流、滴加、搅拌的标准反应装置,置于-10℃的冷水浴中。将环己二氯磷(0.03mol,5.56g)的40ml乙腈溶液,三乙胺(0.07mol,7g)加入到三口烧瓶中,氮气氛围下,当温度稳定在-10℃时,在机械搅拌下逐滴滴加全氟辛基丙醇(0.028mol,13.40g),滴加完成后,此温度下继续搅拌30min。再逐滴滴加丙烯酸羟乙酯(0.035mol,5g),滴加过程控制温度-10℃-5℃。完全滴加完成后,缓慢升温到75℃左右,保温反应4h。反应结束后冷却,过滤除去三乙胺盐酸盐,滤液减压蒸除溶剂和过量三乙胺,得化合物m4,稍微过量的丙烯酸羟乙酯可以作为活性稀释剂在体系中共同使用。核磁数据表征为1hnmr(cdcl3,400mhz):δ6.41(d,1h),6.12(q,1h),5.83(d,1h),4.29(t,2h),3.81(m,4h),1.55(m,8h),1.43(m,4h),1.3(m,2h).核磁表明成功合成了对应化合物m4。实施例5本实施例提供一种述氟碳链改性亚磷酸酯类丙烯酸酯化合物,为亚磷酸酯丙烯酸酯类单体,其结构式如下:该单体通过如下方法制备得到。取带有回流、滴加、搅拌的标准反应装置,置于-10℃的冷水浴中。将(三氟甲基)二氯化磷(0.03mol,5.13g)的40ml乙腈溶液,三乙胺(0.07mol,7g)加入到三口烧瓶中,氮气氛围下,当温度稳定在-10℃时,在机械搅拌下逐滴滴加全氟辛基丙醇(0.028mol,13.40g),滴加完成后,此温度下继续搅拌30min。再逐滴滴加丙烯酸羟乙酯(0.035mol,5.21g),滴加过程控制温度-10℃-5℃。完全滴加完成后,缓慢升温到75℃左右,保温反应4h。反应结束后冷却,过滤除去三乙胺盐酸盐,滤液减压蒸除溶剂和过量三乙胺,得化合物m5,稍微过量的丙烯酸羟乙酯可以作为活性稀释剂在体系中共同使用。核磁数据表征为1hnmr(cdcl3,400mhz):δ6.42(d,1h),6.12(q,1h),5.84(d,1h),4.29(t,2h),3.80(m,4h),1.57(m,2h),1.50(m,2h).核磁表明成功合成了对应化合物m5。实施例6本实施例提供一种述氟碳链改性亚磷酸酯类丙烯酸酯化合物,为亚磷酸酯丙烯酸酯类单体,其结构式如下:该单体通过如下方法制备得到。取带有回流、滴加、搅拌的标准反应装置,置于-10℃的冷水浴中。将二氯(全氟苯基)化磷(0.03mol,8.12g)的40ml乙腈溶液,三乙胺(0.07mol,7g)加入到三口烧瓶中,氮气氛围下,当温度稳定在-10℃时,在机械搅拌下逐滴滴加全氟辛基丙醇(0.028mol,13.50g),滴加完成后,此温度下继续搅拌30min。再逐滴滴加丙烯酸羟己酯(0.035mol,5.21g),滴加过程控制温度-10℃-5℃。完全滴加完成后,缓慢升温到75℃左右,保温反应4h。反应结束后冷却,过滤除去三乙胺盐酸盐,滤液减压蒸除溶剂和过量三乙胺,得化合物m6,稍微过量的丙烯酸羟乙酯可以作为活性稀释剂在体系中共同使用。核磁数据表征为1hnmr(cdcl3,400mhz):δ6.48(d,1h),6.40(q,1h),3.97(t,2h),3.80(m,4h),2.01(s,3h),1.57(m,4h),1.52(m,4h),1.43(m,4h).核磁表明成功合成了对应化合物m6。实施例7~12和对照例1~4将实施例1~6得到的单体m1~m6作为助剂,与助剂ultranox626、hp-10、etanox398对比。其中各商业化的亚磷酸酯类抗氧剂的牌号以及结构如下:实施例4~6和对比例1~4分别选用不同的引发剂体系(如表1)来制备光固化材料,具体过程如下:取季戊四醇四丙烯酸酯di-tmpta50g、活性稀释剂己二醇二丙烯酸酯hdda50g、六官能聚氨酯丙烯酸酯b605(广东博兴新材料有限公司)150g,光引发剂2.5g(1wt%),助剂2.5g(1wt%)搅拌均匀,线棒涂膜玻璃板上,控制膜厚约25μm。以1000w中压汞灯(辐照光强24.9mw/cm2)进行辐照,辐照时间10sec。以指触法检测表面固化情况,涂层表面出现指纹压痕,以×表示,表明存在显著表面氧阻聚;涂层表面未出现指纹压痕,以⊙表示,表明光固化表面氧阻聚得到克服。测试结果列于表1。结果表明本发明制备的亚磷酸酯类丙烯酸酯类单体可有效解决表面氧阻聚问题。表1实施例7~12和对比例1~4提供的光固化材料的性能测试结果样品光引发体系指触法结果实施例71wt%tpo+1wt%m1⊙实施例81wt%tpo+1wt%m2⊙实施例91wt%tpo+1wt%m3⊙实施例101wt%tpo+1wt%m4⊙实施例111wt%tpo+1wt%m5⊙实施例121wt%tpo+1wt%m6⊙对照例11wt%tpo+1wt%ultranox626×对照例21wt%tpo+1wt%hp-10⊙对照例31wt%tpo+1wt%etanox398×对照例41wt%tpo×以上所述是本发明的特定示例实施方式,对于本领域的技术人员,在不脱离本发明的原理下,还可以做出若干的改进与修辞。事实上,本发明的范围由所附的权利要求及其等效限定。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1