头孢氨苄杂质的合成方法与流程
本发明涉及有机合成技术领域,特别涉及一种头孢氨苄杂质的合成方法。
背景技术:
头孢氨苄是第一代可用于口服的头孢类抗生素,1967年由美国礼莱公司研究开发,并在1970年首次投放市场。头孢氨苄主要通过抑制细胞壁的合成,从而使细胞内容物过度生长膨胀至破裂,导致细胞内容物外泄,杀死细菌。其具有抗菌谱广、杀菌力强、耐酸、胃肠吸收好等优点,口服吸收后分布良好,一小时可达血药浓度峰值,但吸收量不减,能投入各种组织中。该药主要用于治疗呼吸道感染、尿路感染和皮肤软组织感染等。
药物的质量是衡量药物品质的重要标准,药物的质量首先决定于药物自身的疗效和毒副作用,即药物的有效性和安全性。因此要求药物在治疗的范围内,不产生严重的毒性反应,不产生或较少产生副作用。药物的有效成分的含量是反映药物纯度的重要标志,而药物中存在的杂质直接影响到药物的疗效并可能导致非治疗活性的毒副作用,必须加以控制。
通过研究发现,头孢氨苄在生产过程中会产生一种特定的杂质(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,cas号为146794-70-9,该头孢氨苄杂质的结构式如下,且至今尚无文献报导其合成方法。
为了控制药物质量,在药品注册申报过程中对该头孢氨苄杂质有明确要求,不过,目前国际上通行的方法是对药品中该杂质进行杂质对照品的研究分析验证。与此同时,为了保证药物的安全性,也需要对该头孢氨苄杂质进行毒理研究,具体包括对其毒性和可能产生的副作用进行研究评估。但由于该头孢氨苄杂质为特定杂质,且市场上也鲜有出售该杂质,因此基于该特定杂质对头孢氨苄药物的质量控制、安全评估有着重要的价值,所以研究该头孢氨苄杂质的合成便有着非常重要的现实意义。
技术实现要素:
有鉴于此,本发明旨在提出一种头孢氨苄杂质的合成方法,以能够进行前述头孢氨苄杂质的合成。
为达到上述目的,本发明的技术方案是这样实现的:
一种头孢氨苄杂质的合成方法,该方法包括如下的步骤:
a、将7-氨基去乙酰氧基头孢烷酸与四甲基胍反应得到7-氨基去乙酰氧基头孢烷酸盐;
b、将7-氨基去乙酰氧基头孢烷酸盐与特戊酰氯进行反应得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸盐;
c、将稀盐酸与步骤b所得产物充分混合,进行水解反应,反应结束后静置分层,取下层有机相并将所取有机相减压蒸发结晶,以得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸。
进一步的,步骤a为在0~10℃下进行反应,步骤b为在0~25℃下进行反应,步骤c为在15~40℃下进行反应。
进一步的,步骤a中的7-氨基去乙酰氧基头孢烷酸与四甲基胍的摩尔比为1:(1~1.5)。
进一步的,步骤a中的反应时间为0.5~1小时。
进一步的,步骤a和b中的反应所用的溶剂为二氯甲烷、三氯甲烷和dmf中的一种或两种。
进一步的,步骤b中的7-氨基去乙酰氧基头孢烷酸盐与特戊酰氯的摩尔比为1:(0.5~1.5)。
进一步的,步骤b中的反应时间2~5小时。
进一步的,步骤c中稀盐酸的质量浓度为1%~15%。
进一步的,步骤c中所用稀盐酸质量浓度为2.5%~10%,且所加入的稀盐酸中所含有的氯化氢与步骤b所得产物的摩尔比为1:(1.0~2.0)。
进一步的,步骤c中水解反应的温度为25~35℃,水解反应的时间为10~60分钟。
相对于现有技术,本发明具有以下优势:
本发明的合成方法采用7-氨基去乙酰氧基头孢烷酸、四甲基胍及特戊酰氯和稀盐酸,且经由水解、静置及减压蒸发结晶,可制得头孢氨苄杂质,而为头孢氨苄杂质的获得提供了一种行之有效的方法。
本发明步骤a中的四甲基胍起到羧基保护作用,可有效地避免后续步骤中羧基参与反应,以减少副产物的生成。步骤b中7-氨基去乙酰氧基头孢烷酸盐与特戊酰氯的反应为胺的亲核进攻反应,以通过胺上的氮原子所含有一对孤对电子进攻酰氯的羰基碳,从而脱掉一分子氯化氢以得到酰胺。而步骤c中的反应则为水解反应,以脱除与羧基成盐的四甲基胍,进而得到最终的合成品。
本发明的合成方法反应原料相对易得,反应过程操作简单,反应设备要求低,且反应条件相对温和,合成产物含量高,在目前该头孢氨苄杂质市场价格较高且难以买到的情况下,为头孢氨苄杂质研究提供了相对便捷可靠的获得渠道,并大大降低了其成本,对于更深入广泛的研究头孢氨苄相关用药安全性、可靠性、稳定性以及生产过程中的质量控制有着很大的促进作用。
附图说明
构成本发明的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
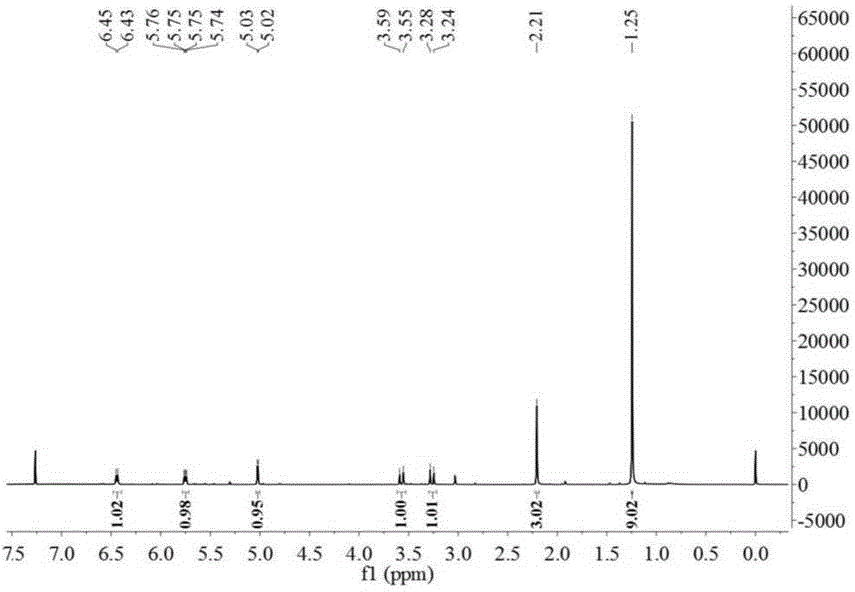
图1为本发明实施例所合成的头孢氨苄杂质的氢核磁谱图。
具体实施方式
需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
本实施例涉及头孢氨苄杂质的合成方法,该头孢氨苄杂质具体为(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,且该合成方法包括:
步骤a、在0~10℃下,将摩尔比为1:(1~1.5)的7-氨基去乙酰氧基头孢烷酸与四甲基胍反应0.5~1小时得到7-氨基去乙酰氧基头孢烷酸盐。
步骤b、在0~25℃下,将摩尔比为1:(0.5~1.5)的7-氨基去乙酰氧基头孢烷酸盐与特戊酰氯进行反应2~5小时得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸盐。
步骤c、在15~40℃下,将质量浓度为1%~15%的稀盐酸与步骤b所得产物充分混合,在25~35℃下进行10~60分钟的水解反应,水解反应结束后静置分层,取下层有机相并将所取有机相减压蒸发结晶,即得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,也即所述的头孢氨苄杂质。
其中,步骤a和步骤b中的反应所用的溶剂为二氯甲烷、三氯甲烷和dmf中的一种或两种。而步骤c中所用稀盐酸质量浓度优选的为2.5%~10%,同时所加入的稀盐酸中所含有的氯化氢与步骤b所得产物的摩尔比也为1:(1.0~2.0)。
本实施例的头孢氨苄杂质的合成过程由结构式表达如下:
而下面则以若干具体的制备实例进一步说明本实施例的头孢氨苄杂质的合成。
制备实例1
本实例的头孢氨苄杂质的合成包括的步骤如下。
步骤a、在干燥洁净的反应瓶中,加入40ml二氯甲烷和5g7-氨基去乙酰氧基头孢烷酸,降温至0℃,向反应瓶中加入2.8g四甲基胍,控制反应温度不高于10℃,反应30分钟,得到7-氨基去乙酰氧基头孢烷酸盐溶液。
步骤b、在控温不超过10℃下向反应瓶中加入3g特戊酰氯,加料结束后,控制温度在0℃,反应4小时。
步骤c、向水解反应瓶中加入质量浓度为5.64%的盐酸70ml,调节温度至30℃,将步骤b反应结束后的产物倒入水解反应瓶中充分搅拌30分钟,静置30分钟,取下层有机相,将有机相进行减压蒸发结晶得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸4.6801g。测得其收率为67.21%,纯度为98.07%,且其核磁结构鉴定结果如下:
1hnmr(500mhz,cdcl3)δ:6.45~6.43(d,j=8.3hz,1h),5.76~5.73(dd,j=8.3hz,j=4.6hz,1h),5.03~5.02(d,j=4.6hz,1h),3.59~3.55(d,j=18.6hz,1h),3.24~3.28(d,j=18.6hz,1h),2.21(s,2h),1.25(s,9h)。
制备实例2
本实例的头孢氨苄杂质的合成包括的步骤如下。
步骤a、在干燥洁净的反应瓶中,加入10ml二氯甲烷、30ml三氯甲烷和5g7-氨基去乙酰氧基头孢烷酸,降温至4℃,向反应瓶中加入3.2g四甲基胍,控制反应温度不高于10℃,反应40分钟,得到7-氨基去乙酰氧基头孢烷酸盐溶液。
步骤b、在控温不超过10℃下向反应瓶中加入2g特戊酰氯,加料结束后,控制温度在4℃,反应3小时。
步骤c、向水解反应瓶中加入质量浓度为2.84%的盐酸70ml,调节温度至35℃,将步骤b反应结束后的产物倒入水解反应瓶中充分搅拌40分钟,静置30分钟,取下层有机相,将有机相进行减压蒸发结晶得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸2.5822g。测得其收率为51.95%,纯度为98.52%,且其核磁结构鉴定结果如下:
1hnmr(500mhz,cdcl3)δ:6.45~6.43(d,j=8.3hz,1h),5.76~5.73(dd,j=8.3hz,j=4.6hz,1h),5.03~5.02(d,j=4.6hz,1h),3.59~3.55(d,j=18.6hz,1h),3.24~3.28(d,j=18.6hz,1h),2.21(s,2h),1.25(s,9h)。
制备实例3
本实例的头孢氨苄杂质的合成包括的步骤如下。
步骤a、在干燥洁净的反应瓶中,加入20ml二氯甲烷、20mldmf和5g7-氨基去乙酰氧基头孢烷酸,降温至7℃,向反应瓶中加入4g四甲基胍,控制反应温度不高于10℃,反应50分钟,得到7-氨基去乙酰氧基头孢烷酸盐溶液。
步骤b、在控温不超过10℃下向反应瓶中加入3.4g特戊酰氯,加料结束后,控制温度在9℃,反应2小时。
步骤c、向水解反应瓶中加入质量浓度为7%的盐酸70ml,调节温度至25℃,将步骤b反应结束后的产物倒入水解反应瓶中充分搅拌50分钟,静置30分钟,取下层有机相,将有机相进行减压蒸发结晶得到(6r,7r)-7-[(2,2-二甲基-1-氧代丙基)氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸5.0118g。测得其收率为71.84%,纯度为98.26%,且其核磁结构鉴定结果如下:
1hnmr(500mhz,cdcl3)δ:6.45~6.43(d,j=8.3hz,1h),5.76~5.73(dd,j=8.3hz,j=4.6hz,1h),5.03~5.02(d,j=4.6hz,1h),3.59~3.55(d,j=18.6hz,1h),3.24~3.28(d,j=18.6hz,1h),2.21(s,2h),1.25(s,9h)。
本实施例合成的头孢氨苄杂质的氢核磁谱图如图1中所示,而由以上各实例可以看出由本实施例的合成方法所合成的头孢氨苄杂质的纯度大于98%,其可很好的满足头孢氨苄杂质的研究,而有着很好的现实意义。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!