一种将寡聚核苷酸逆转录成cDNA的方法及寡聚核苷酸检测方法与流程
本发明涉及rna检测技术领域。具体地说是一种将寡聚核苷酸逆转录成cdna的方法及寡聚核苷酸检测方法。
背景技术:
生物体内天然存在片段长度小于10个nt的寡聚核苷酸,如转录起始时产生的流产性转录本(abortivetranscript,at)和长片段核苷酸降解后产生的小片段核苷酸)。现有发现暗示这些小片段核苷酸在体内可能有重要的生物学作用,但对10nt以下的小片段核苷酸一直无法进行定性和定量检测。
技术实现要素:
为此,本发明所要解决的技术问题在于提供一种将寡聚核苷酸逆转录成cdna的方法及寡聚核苷酸检测方法,不仅能够检测10nt以上的核苷酸,特别是能够检测6-10nt长的小片段核苷酸。
为解决上述技术问题,本发明提供如下技术方案:
一种将寡聚核苷酸逆转录成cdna的方法,包括如下步骤:
(1)设计单链dna,所述单链dna的中间部分与目标寡聚核苷酸碱基互补;
(2)根据目标寡聚核苷酸选择上游辅助rna和下游辅助rna,且所述上游辅助rna的3’端含有单羟基,所述下游辅助rna的5’端含有单磷酸基团;所述单链dna的两端分别与所述上游辅助rna和所述下游辅助rna碱基互补;
(3)将目标寡聚核苷酸两端分别与所述上游辅助rna和所述下游辅助rna连接起来;
(4)将所述单链dna消化掉,得到中间部分为目标寡聚核苷酸的待逆转录单链rna;
(5)在待逆转录单链rna的3’端设计引物,将待逆转录单链rna逆转录成cdna。
上述将寡聚核苷酸逆转录成cdna的方法,在步骤(3)中:用连接酶将目标寡聚核苷酸两端分别与所述上游辅助rna和所述下游辅助rna连接起来。
上述将寡聚核苷酸逆转录成cdna的方法,在步骤(3)中:所述连接酶为t4dna连接酶。
上述将寡聚核苷酸逆转录成cdna的方法,在步骤(1)中:用dnasei将所述单链dna消化掉。
寡聚核苷酸检测方法,包括如下步骤:
(a)按照权利要求1-6任一所述的方法,将寡聚核苷酸逆转录成cdna;
(b)对cdna进行qpcr定量检测、高通量测序检测或基因芯片检测,从而可以对体内产生的寡聚核苷酸进行qpcr定量检测、高通量测序检测或基因芯片检测。
上述寡聚核苷酸检测方法,在步骤(b)中,qpcr定量检测方法如下:
第一种方法:根据cdna设计上游引物primerf和下游引物primerr,上游引物primerf与待逆转录单链rna的上游端相同,下游引物primerr与待逆转录单链rna的下游端碱基互补,然后进行实时定量pcr检测;
第二种方法:设计taqman-mgb探针,taqman-mgb探针覆盖待逆转录单链rna中目标寡聚核苷酸及其两端若干个碱基序列,用高特异的taqman探针pcr法检测目标寡聚核苷酸的含量。
上述寡聚核苷酸检测方法,在步骤(b)中:高通量测序检测方法如下:
(b1-1)根据cdna设计上游引物primerf和下游引物primerr,然后用上游引物primerf和下游引物primerr对cdna进行第一轮pcr;
(b1-2)设计包含primerf的上游引物,其包括用于和illumina测序仪结合和cluster形成的p5序列以及用于read1的测序的i5序列,i5序列位于p5序列和primerf之间;同时设计包含primerr的下游引物,其包括用于和illumina测序仪结合和cluster形成的p7序列、用于read2的测序的i7序列、以及位于p7序列和i7序列之间的barcode序列,并且i7序列位于barcode序列和primerr之间;barcode序列用来区分样品。
上述寡聚核苷酸检测方法,在步骤(b)中,基因芯片检测方法如下:在上游辅助rna和下游辅助rna上设计限制性核酸内切ecorv的酶切位点,在完成pcr扩增后,用ecorv处理pcr产物,就会产生包含目标寡聚核苷酸序列的dna片断,然后设计相应的探针,最后进行检测。
本发明的技术方案取得了如下有益的技术效果:本发明的通过设计一条单链dna,其中间部分与寡聚核苷酸互补,两端分别与上游辅助rna和下游辅助rna互补;且上游辅助rna3’端有羟基,下游辅助rna的5’端含有一个磷酸基团。然后用t4dna连接酶将寡聚核苷酸与辅助rna连接起来,接下来用dnasei将dna链消化掉,然后在新连接rna的3’端设计引物,将新连接的rna链逆转录成cdna,从而可以对体内产生的6nt以上的寡聚核苷酸进行实时定量pcr检测、测序检测或芯片检测。现有技术中长度大于10nt的核苷酸已经有方法可以检测,本发明最大特征是可以检测6-10nt长的寡聚核苷酸。
附图说明
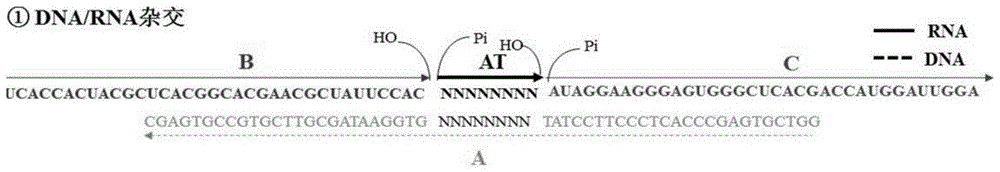
图1dna/rna碱基堆积杂交示意图;
图2t4dna连接酶连接;
图3dnasei消化a链;
图4cdna合成;
图5taqman-mgb探针实时定量pcr检测;
图6芯片法检测6-10nt寡聚核苷酸;
图7muc168ntat探针示意图;
图8反转录示意图;
图9实施例2中的电泳图;
图10sanger测序结果。
具体实施方式
实施例1
本发明可用于检测生物体内天然存在的核苷酸降解后产生的6nt以上的小片段核苷酸,也可以用于检测流产性转录本,以7nt的at为例,详解如下:
1、rna提取与准备
因为寡聚核苷酸的长度较短,所以其提取方法与常规的rna提取方法略有不同。另外在提取以后如果要检测的对象是at,还要先将5′端只有一个磷酸基团的长链rna去除磷酸基团,从而使所有降解产生的寡聚核苷酸不能在后续步骤中被连接起来,然后再将5′端有3个磷酸的流产性转录本去掉两个磷酸基团,以便下一步只将at与辅助rna连接起来。在下述方法中,如果要检测体内所有寡聚核苷酸,则不进行第9步操作;如果只检测rna降解后产生的10nt以下的小片段核苷酸,则不进行第9和第10步操作;如果只想检测at,则需要进行等比全部操作。
(1)在样品中加入1.5x体积的trizol,混匀,室温静置裂解15min;
(2)加入200ul/mltrizol-氯仿,颠倒混匀15s,室温静置5min;
(3)12000rpm/4℃,15min,离心,保留上清;
(4)量取上清液的体积,缓慢加入上清液体积0.43倍的无水乙醇(500μl的转移液加215μl无水乙醇),混匀(此时可能会出现沉淀)。将得到的溶液和沉淀一起转入向吸附柱mirspin中,室温12,000rpm离心30sec,若一次不能将全部溶液和混合物加入向吸附柱mirspin中,分两次转入,离心后弃掉向吸附柱mirspin,保留流出液;
(5)向流出液中加入1/10体积的3mnaac,缓慢混匀;缓慢加入1.8倍的无水乙醇[使其最终浓度为75%],混匀;
(6)-80℃,静置沉淀36h;
(7)12000rpm/4℃,30min,弃上清;
(8)晾干,加入30ulrnase-freeh2o溶解;
(9)在溶解液中加入热敏小牛肠碱性磷酸酶(cip)(除去小rna中可能存在的rna裂解片段5′端的磷酸单脂),37℃孵育10分钟,然后加热到80℃并保持2分钟,以失活cip;
(10)在上一步产物中加入rna5’-polyphosphatase(此酶可以专一从有3个磷酸基团的rna5′端降解掉2个磷酸,只保留1个单磷酸)和5’-polyphosphatase专用buffer,37℃孵育30分钟。然后加热到65℃并保持30分钟,以失活rna5’-polyphosphatase。
2、cdna合成
(1)dna-rna杂交:6-10nt寡核苷酸因为链太短,无法与探针链(a)牢固结合,但如果在其上下游已经有辅助rna链(b和c,各长35bp,且序列在待检样品所属物种中具有高度特异性)稳定结合在探针上,则可以与探针形成稳定结合(碱基堆积杂交原理)。在本实验中,at与探针链a中间(8个n所示区)完全互补,b和c则与其上下游互补,可以如图1所示杂交,形成稳定的杂交链。
(2)rna-nick连接:t4dnaligase可以修补dna/rna杂合物上的单链缺刻(single-strandnicks),利用其活性催化dna-rna杂交处的rna-nick(3’-oh+5’-pi),形成一条b+d+c的完整的rna(e),e与a有52个bp互补。
(3)dnasei消化a链:dsn是一种热稳定核酸酶,该酶能够选择性降解双链dna和dna-rna杂交体中的dna,但对单链核酸分子几乎没有作用。降解aprobe仅保留e(rna)链(如图3所示)。
(4)e(rna)反转录:根据c的序列,在e链近3’端处设计链特异性引物(spr)进行反转录(如图4所示)。
3、qpcr定量检测(如图5所示)
设计上下游引物(primerf和primerr),进行实时定量pcr检测,为了提高特异性,也可设计覆盖d序列的taqman-mgb探针,用高特异的taqman探针pcr法检测6-10nt寡核苷酸的含量。
4、高通量测序
用3中提到的上游引物primerf和primerr对合成的cdna进行第一轮pcr,以对目标序列进行扩增,然后设计包含primerf的上游引物,其中有下划横线为:p5序列,用于和illumina测序仪结合和cluster形成;方框内为:i5序列,用于read1的测序;
同时设计包含primerr的下游引物,有下划横线为:p7序列,用于和illumina测序仪结合和cluster形成;方框内为:i7序列,用于read2的测序;中间的barcode序列用来区分样品,可选的有很多种(其中几种列举如下):1)cgtgat、2)acatcg、3)gcctaa、4)tggtca、5)cactgt、6)attggc、7)gatctg、8)tcaagt。
5、基因芯片检测
用本方法获得的cdna也可用芯片法进行检测,如下图所示,只需在辅助rna链b和c合适位置设计限制性核酸内切ecorv的酶切位点(图6中蓝色字母所示),在完成pcr扩增后,用ecorv处理pcr产物,就会产生包含at序列的24nt的dna片断,然后就可设计相应的探针,进行检测了。如果我们检测7nt长的寡聚核苷酸的话,只要设计47个探针,也就是16384个探针,就可以检测所有的7nt长的寡聚核苷酸了。
实施例2
本实施例利用实施例1中的方法检测a549/hela细胞(本实验室保存)中ca125原始基因muc16的8ntat和人工合成的同序列8ntat(5’端加单磷酸基团)的含量,其过程和结果如下。
1、细胞培养
(1)用75cm2培养瓶(全培养基rpmi1640+1%penicillin/streptomycin+10%fbs)在37℃、5%co2条件下培养细胞至90%左右;
(2)1xpbs清洗细胞后,用3mltrypsin-edtasolution消化1分钟,加3ml全培养基终止反应;
(3)1000rpm离心3分钟收集细胞,重悬计数。
2、samllrna提取
(1)收集的细胞中加入3ml裂解液mz(buffermz,tiangendp501),上清液中等体积加入buffermz,充分混匀,室温裂解15分钟;
(2)加入200ul/1mltrizol氯仿,颠倒混匀15s,室温静置5分钟;
(3)12000rpm,4℃离心15分钟,保留上清;
(4)量取转移液的体积,缓慢加入转移液体积0.43倍的无水乙醇(500μl的转移液加215μl无水乙醇),混匀(此时可能会出现沉淀)。将得到的溶液和沉淀一起转入向吸附柱mirspin(tiangendp501)中,室温12000rpm(~13,400×g)离心30sec,分两次将全部溶液和混合物加入向吸附柱mirspin中,转入,离心后弃掉向吸附柱mirspin,保留流出液;
(5)向滤液中加入1/10体积的3mnaac,缓慢混匀并加入1.8倍的无水乙醇,混匀;
(6)-80℃,静置沉淀36h;
(7)12000rpm,4℃离心30分钟,弃上清;
(8)用80%的无水乙醇洗涤两次;
(9)晾干,加30ulrnase-freeh2o溶解,备用。
3、rna处理
(1)磷酸单脂去磷酸化用重组表达的热敏小牛肠碱性磷酸酶(cip)(neb,货号m0525)完成rna5′-磷酸单脂的去磷酸化反应。
①在20μl反应体系中加入1pmol的rna,10xcutsmartbuffer2μl,quickcip1μl,然后补rnasefreeh2o至20μl。
②37℃孵育10分钟,然后加热到80℃并保持2分钟。
(2)去除at5’端的γ,β磷酸基团
用rna5’-polyphosphatase(epicentre,货号rp8092h)去除at5’端的γ,β磷酸基团。
①rnasefree离心管中加入10x5’-polyphosphatasereactionbuffer2μl,
riboguardrnaseinhibitor0.5μl,rna5’-polyphosphatase(20units)1μl,
rna5′-磷酸单脂的去磷酸化反应液补至20μl;
②37℃孵育30分钟,然后加热到65℃并保持20分钟使酶失活。
4、杂交检测
(1)、dna/rna杂交muc16的前8个起始转录序列为aagcgttg,我们设计了下图7所示的探针和辅助序列检测8nt的流产性转录本,杂交步骤如下:
①体系5xt4-dnaligasebuffer4μl,a链、b链和c链各4μl(1μm),rnasample4μl或人工合成at(1μm),缓慢混匀。
②退火90℃孵育2分钟、85℃孵育2分钟、80℃孵育2分钟、75℃孵育2分钟、70℃孵育2分钟、65℃孵育2分钟、60℃孵育5分钟、55℃孵育5分钟、50℃孵育5分钟、45℃孵育5分钟、40℃孵育5分钟、35℃孵育5分钟、30℃孵育5分钟、25℃孵育30分钟。
(2)、t4-dnaligase连接在上步反应体系中直接加入t4-dnaligase(no.15224-017invitrogen)2.2μl,14℃,24小时连接。
(3)、用dsn(duplex-specificnuclease)消化反应体系中的dna10xdsnbuffer1ul,dsn(ea001evrogen)0.6μl,t4-dnaligase连接产物8.4ul。60℃反应2h,92℃10分钟。
(4)、cdna反转录(rr0047takara)图8所示
反应体系:primescriptrtenzymemixi1ul,5×primescriptbuffer24ul,specificityprimer(1um)1ul,rna样品10ul,rnasefreeh2o至20μl。42℃反应20分钟,85℃反应5秒使酶失活,备用。
(5)、pcr扩增(takararr003)
premixtaq(extaqversion2.0)10ul,primerf(10um)1ul,specificity(10um)1ul,cdna200ng,ddh2o补齐至20ul。94℃1分钟,94℃20s/68℃1分钟35xcycles,72℃5分钟。
(6)、聚丙烯酰胺凝胶电泳鉴定(12%)
40%丙烯酰胺3ml,5xtbe1ml,10%aps50ul,temde10ul,ddh2o补齐至10ml。图9电泳结果如图,其中1道为人工合成的at,2道为从细胞中提取的rna得到的结果;
(7)、测序:在上下游引物的5’-end各加10个t延长pcr产物,电泳检测确定产物大小,切胶回收后送北京睿博兴科生物技术有限公司进行sanger测序,结果如下[图10],因sanger测序技术的限制,5’端缺失了22个bp,但余下测序序列与5’端辅助链后12个序列、目标流产性转录本序列,3’端辅助链的序列完全匹配,证明实验结果可靠。
由实施例2的实际检测结果证明,本发明的方法是可行的。
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本专利申请权利要求的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!