图灵结构共价有机框架膜材料及其用途
1.本发明属于有机脱酸材料领域,具体涉及图灵结构共价有机框架膜材料及其用途。
背景技术:
2.自然界处处可见纷繁复杂、精妙绝伦的图案与形态,例如动物的斑纹、植物的轮状叶序等,科学家图灵用一个“反应
‑
扩散”方程揭示了这些自然形态的化学本质,即某些重复的自然斑图可能由两种特定物质(分子、细胞等)相互反应或作用而产生1。后来,科学家们将这种由两种组分自发组织成的斑纹、条纹、环纹、螺旋,或是斑驳的斑点等结构称为“图灵结构”2
‑4。图灵结构的产生一般需要在反应
‑
扩散体系中进行,界面聚合反应体系就是这样一种在非热力学平衡状态下的反应扩散体系5‑7。由界面聚合反应制备的具有图灵结构的膜在透水性和水盐选择性方面具有出色的传输性能,这种特性使得这类膜材料在膜分离领域有着巨大的应用潜力8‑
11
。然而,目前对于图灵结构膜材料的相关研究仍然非常少见。因此,研究新型图灵结构膜材料并开发其在膜分离领域中的巨大应用价值有着重要的科学意义。
3.膜分离技术是指利用不同组分在膜上选择性透过能力的差异,以外界条件(压力、电场、热等)或者组分浓度差为驱动力,对组分进行选择性分离、提纯的方法
12
‑
18
。新兴的膜分离技术由于其特有的能耗低、分离过程简单、材料可重复利用等特点在许多领域得到了广泛的应用
19
‑
22
。尤其是面对乏燃料后处理过程中产生的料液时,膜分离方法更能充分发挥其技术优势
23
‑
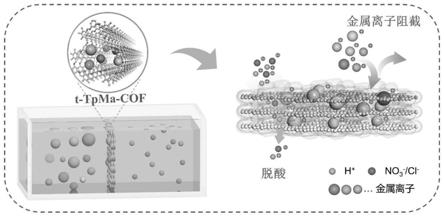
25
。后处理产生的料液的酸度一般在3mol/l以上,通常需要采用煅烧脱硝、稀释或者中和的方法以降低溶液酸度
26,27
。煅烧不仅需要很高的温度,而且在煅烧过程中,高温下挥发的硝酸对尾气设备有很强的腐蚀,料液中的裂变产物ru、tc等在高酸度的溶液中也易挥发出来,造成极大的不利影响
28
‑
30
。稀释会使废液的体积增大,而中和则会增加溶液中的盐含量,这都会大大增大废物处理的难度、处理量和处置费用
31
。膜分离方法改变了传统的脱酸思路,直接通过质子传输降低料液酸度,打破了对重金属敏感、耗能高、脱酸过程产生挥发气体等限制,在乏燃料后处理过程的极端环境下为料液的脱酸提供了简便、灵活的新方法。然而,面对具有放射性水平高、衰变热功率和生物毒性大、化学成分复杂、酸性强、腐蚀性大等特点的料液时,许多膜材料均难以发挥出其在分离方面的优势
32
。因此,研究与开发能够适用于乏燃料后处理真实料液环境的膜分离材料,仍然是研究人员关注的焦点。
技术实现要素:
4.为解决上述问题,本发明提供了一种图灵结构共价有机框架膜材料。
5.上述图灵结构共价有机框架膜材料,采用有机
‑
有机溶剂界面法,以2,4,6
‑
三羟基
‑
1,3,5
‑
苯三甲醛为节点单体与间苯二胺通过醛胺缩合反应,合成得到图灵结构共价有机框架(t
‑
tpma
‑
cof)膜材料。
6.上述图灵结构共价有机框架膜材料的制备方法,其合成示意图如图1所示。
7.上述图灵结构共价有机框架膜材料的制备方法,包括以下步骤:将tp(2,4,6
‑
三羟基
‑
1,3,5
‑
苯三甲醛)溶于下层溶剂中,加入缓冲层;再将ma(间苯二胺)溶解在上层溶剂中,将其在缓冲层的顶部持续缓慢加入;在常温不受干扰条件下放置7天;缓冲层消失,在上层和下层的界面处得到t
‑
tpma
‑
cof膜材料,分别用水和ch2cl2(二氯甲烷)洗涤。
8.上述图灵结构共价有机框架膜材料的制备方法中,所述的下层溶剂为ch2cl2。
9.上述图灵结构共价有机框架膜材料的制备方法中,所述的缓冲层为乙酸、sc(otf)3(三氟甲烷磺酸钪)、eu(otf)3(三氟甲烷磺酸铕)或水中的至少一种。所述乙酸的浓度为2~6m。
10.上述图灵结构共价有机框架膜材料的制备方法中,所述的上层溶剂为乙醇、水或ch2cl2与dmf(n,n
‑
二甲基甲酰胺)的混合溶剂。所述ch2cl2与dmf的体积比为6∶1~4。
11.上述图灵结构共价有机框架膜材料的制备方法中,所述ma溶于上层溶剂的浓度为0.5~2mmol/l。
12.上述图灵结构共价有机框架膜材料的制备方法中,所述下层溶剂、缓冲层和上层溶剂的体积比为2∶1~1.5∶1.5~3。
13.本发明还提供了上述图灵结构共价有机框架膜材料在制备脱酸剂中的用途。所述的脱酸剂是用于乏燃料后处理料液的脱酸处理。
14.本发明提供的t
‑
tpma
‑
cof材料拥有良好的热稳定性和酸碱稳定性。在微观结构上,t
‑
tpma
‑
cof拥有两面不同形状的图灵结构,一面为线状图灵结构,一面为孔状的图灵结构。令人惊奇的是,两种结构拥有明显不同的性质,线状图案的那一面展现出疏水的特性,而孔状图案的那一面则展现出超亲水的性质。离子筛分实验中,t
‑
tpma
‑
cof纳滤膜展现出了出色的金属离子阻截和质子透过性能,在酸度高达5m hno3条件下仍能保持对h
+
的选择性透过性能,这使得t
‑
tpma
‑
cof能够轻松适应乏燃料后处理料液的苛刻环境,用于料液的脱酸处理。同时,t
‑
tpma
‑
cof在染料筛分中也有着出色的表现。t
‑
tpma
‑
cof的成功合成与应用,为设计新型图灵结构膜材料提供了可选择的途径,拓宽了具有图灵结构膜材料的应用领域。
附图说明
15.图1图灵结构t
‑
tpma
‑
cof膜材料的合成示意图。
16.图2图灵结构t
‑
tpma
‑
cof膜材料的表征图谱:(a)t
‑
tpma
‑
cof和单体的红外谱图;(b)t
‑
tpma
‑
cof的13c固体核磁谱图;(c)pxrd图;(d)氮气吸附脱附等温线(插图为t
‑
tpma
‑
cof的孔径实验值和模拟值)。
17.图3图灵结构t
‑
tpma
‑
cof膜材料的微观结构:(a
‑
c)t
‑
tpma
‑
cof的球差矫正电镜图(插图为t
‑
tpma
‑
cof模拟的结构图);(c)(d)为选区电子衍射图;(e)为t
‑
tpma
‑
cof模拟的层间距。
18.图4图灵结构t
‑
tpma
‑
cof膜材料的扫描电子显微镜图:(a)t
‑
tpma
‑
cof的截面扫描电镜图;(b)t
‑
tpma
‑
cof膜上表面的扫描电镜图;(c)t
‑
tpma
‑
cof膜上表面的接触角;(d)t
‑
tpma
‑
cof膜上表面的原子力显微镜图;(e)t
‑
tpma
‑
cof膜下表面的扫描电镜图;(f)t
‑
tpma
‑
cof膜下表面的接触角;(g)t
‑
tpma
‑
cof膜下表面的原子力显微镜图。
19.图5图灵结构t
‑
tpma
‑
cof膜材料两个面对水的渗透性能:(a)t
‑
tpma
‑
cof膜亲水面
和疏水面的透水率了;(b)1m hno3体系中的h
+
渗透通量。
20.图6 14种竞争离子在酸性体系中通过cof膜的渗透通量:(a)1m hno3体系;(b)3m hno3体系;(c)5m hno3体系;(d)1m hcl体系;(e)3m hcl体系;(f)5m hcl体系。
21.图7 14种竞争离子在(a)ph=1.8和(b)5m的硝酸体系中,过滤时间为192小时透过膜的渗透通量。
22.图8图灵结构t
‑
tpma
‑
cof膜材料脱酸和金属离子阻截的过滤机理示意图。
23.图9(a)t
‑
tpma
‑
cof的热重曲线;(b)t
‑
tpma
‑
cof辐照前后的红外谱图(c)t
‑
tpma
‑
cof硝酸浸泡前后的红外谱图。
具体实施方式
24.图灵结构共价有机框架膜材料的制备方法,包括以下步骤:
25.a、将tp溶于ch2cl2中,加入缓冲层;所述tp溶于下层溶剂的浓度为0.5~2mmol/l;
26.b、再将ma溶解在上层溶剂中,将其在缓冲层的顶部持续缓慢加入;所述ma溶于上层溶剂的浓度为0.5~2mmol/l;
27.c、在常温不受干扰条件下放置7天;缓冲层消失,在上层和下层的界面处得到t
‑
tpma
‑
cof膜材料,分别用h2o和ch2cl2洗涤。
28.所述下层溶剂、缓冲层和上层溶剂的体积比为2∶1~1.5∶1.5~3。
29.上述图灵结构共价有机框架膜材料的制备方法中,所述的缓冲层为乙酸、sc(otf)3、eu(otf)3或水。所述乙酸的浓度为2~6m。
30.上述图灵结构共价有机框架膜材料的制备方法中,所述的上层溶剂为乙醇、水或ch2cl2与dmf的混合溶剂。所述ch2cl2与dmf的体积比为6∶1~4。
31.实施例1t
‑
tpma
‑
cof膜的制备
32.在烧杯中合成了t
‑
tpma
‑
cof膜:首先,将tp溶于下层溶剂中,加入缓冲层。再将ma溶解在上层溶剂中,在缓冲层的顶部持续缓慢加入。在常温不受干扰条件下放置7天。缓冲层消失,在上层和下层的界面处得到t
‑
tpma
‑
cof膜,用h2o和ch2cl2洗涤。溶剂的选取、及用量如表1所示:
33.表1
[0034][0035]
表1中制备得到t
‑
tpma
‑
cof的表征:从红外光谱图(图2a)上可以看到,t
‑
tpma
‑
cof中tp的c=o(1644cm
‑1)特征吸收峰和ma的
‑
nh2(~3200
‑
3430cm
‑1)特征吸收峰消失,c=c(1574cm
‑1)的特征吸收峰的出现,证明了醛胺缩合反应的成功进行。与红外的结果一致,t
‑
tpma
‑
cof的
13
c固体核磁谱(
13
c solid
‑
state nuclear magnetic resonance,
13
c ssnmr)图中(图2b)在~102ppm也出现了c=c
‑
n特征峰。接着,本发明基于粉末x射线(powder x
‑
ray diffraction,pxrd)数据(图2c),借助materials studio(ms)模拟研究了t
‑
tpma
‑
cof的片层堆叠方式。通过ms模拟发现,aa堆叠方式的模拟值与实验值吻合的较好,t
‑
tpma
‑
cof在9.25
°
和26.82
°
出现的衍射峰可分别对应100,001晶面。一般而言,材料的堆积方式不同,其孔尺寸也会有明显的差别。若采用aa堆叠方式,材料的相应孔径为左右;若采用ab堆叠,材料的孔将会由于交错阻挡而大大减小,为为了进一步确定t
‑
tpma
‑
cof的堆积方式,本发明对t
‑
tpma
‑
cof进行了n2吸附脱附表征。结果如图所示(图2d),t
‑
tpma
‑
cof的吸附等温线呈
ⅰ‑
型吸附等温线形状,说明材料的孔属于微孔。根据非局部密度泛函方法计算可以得出t
‑
tpma
‑
cof的平均孔径为这与aa堆叠的孔径是匹配的,从而证实t
‑
tpma
‑
cof是采用aa堆叠的。
[0036]
球差矫正电镜被用来深入揭示t
‑
tpma
‑
cof膜材料规则有序的微观结构(图3),通过电镜图可以观察到t
‑
tpma
‑
cof膜材料类似蜂窝状的cof结构框架,其中明亮的斑点和黑色对应于结构单元和1d孔道,这与模拟的结构类型是一致的。电镜图中的晶格纹间距经测量约为0.33nm,这与xrd中在26.82
°
出现衍射峰吻合,此峰对应于t
‑
tpma
‑
cof的001晶面。此外,选定区域电子衍射(saed)图(图3d)显示t
‑
tpma
‑
cof具有明显的电子衍射点。另外,本发明通过元素分析测定了t
‑
tpma
‑
cof中各元素的含量,测量结果与理论计算值较为接近。
[0037]
本发明采用液
‑
液界面聚合法制备的t
‑
tpma
‑
cof膜材料,在两种溶剂的界面处形成了不同的膜表面形貌。在两相界面的水侧形成孔状图灵结构,水接触角测试显示这一侧为超亲水(图4b、c)。在二氯甲烷一侧,形成了类似荷叶表面的密集的线状图灵结构。测量这一侧的接触角为100
°
,表明它是疏水的(图4e、f)。采用扫描电子显微镜(scanning electron microscopy,sem)观察t
‑
tpma
‑
cof膜材料的表面形貌。从sem图像可以看出,亲水的孔状图灵结构由球形团聚体组成(图4b),疏水线型图灵结构由丝状聚集体组成(图4e)。此外,t
‑
tpma
‑
cof膜上下两面不同的形貌特征也可以通过截面sem测试清晰地观察到(图4a)。为了更直观地展现与膜的两面不同形貌直接相关的亲疏水性质,本发明对t
‑
tpma
‑
cof膜进行了一个简单的溶剂驱动的沉浮漂浮实验。将t
‑
tpma
‑
cof膜材料的超亲水面朝上放在水中,t
‑
tpma
‑
cof膜材料会快速的沉入水底,接着本发明将t
‑
tpma
‑
cof膜材料翻转过来,变成疏水面朝上,此时,t
‑
tpma
‑
cof膜材料又会迅速浮上水面。该实验清晰地表明制备的t
‑
tpma
‑
cof膜材料具有一面超亲水一面疏水的两性性质,亲水面与水分子的高度亲和性和疏水面对水分子的排斥特性,造成了膜的快速下沉和上浮现象。另外,本发明使用原子力显微镜(atomic force microscope,afm)对t
‑
tpma
‑
cof膜材料的表面形貌进行了进一步观察。从afm中可以看到(图4d、g),疏水面由于主要由密集的线状结构组成,较为平滑,褶皱和起伏较小,平均表面粗糙度(ra)为215nm,而亲水面主要由疏松的絮状结构组成,起伏极为明显,平均表面粗糙度(ra)为900nm,由此可见t
‑
tpma
‑
cof两面的平均表面粗糙度有很大差别。
[0038]
实施例2t
‑
tpma
‑
cof膜材料的稳定性
[0039]
膜的理化稳定性是保证其在实际环境中有效应用的重要指标之一。考虑到在乏燃料后处理过程中真实料液环境中高温、强辐照和强酸性等严苛的应用条件,本发明对t
‑
tpma
‑
cof膜材料的热稳定性、酸稳定性以及辐照稳定性进行了测试。
[0040]
1)热稳定性:通过n2氛围中的热重分析测量t
‑
tpma
‑
cof膜材料的热稳定性。
[0041]
测量结果表明(图9a),100℃之前的失重是材料物理吸收的水分蒸发引起的。100~400℃之间出现了轻微的质量损失(t
‑
tpma
‑
cof~4.08%),可能是材料中的小分子分解造成的。400℃以上的质量损失明显较快,可能是本身的结构开始分解。达到500℃时还剩余66.8%,表明该材料的热稳定性良好。
[0042]
2)辐照稳定性:对t
‑
tpma
‑
cof膜材料进行的辐照稳定性研究显示(图9b),t
‑
tpma
‑
cof在辐照前后的红外谱几乎没有明显的变化,说明t
‑
tpma
‑
cof具有良好的辐照稳定性,可以耐受105gy的γ射线辐照。
[0043]
上述研究表明t
‑
tpma
‑
cof膜材料有着较高的酸稳定性、热稳定性和辐照稳定性,使其有望应用于乏燃料后处理料液等极端环境中的离子筛分。
[0044]
3)酸稳定性:将t
‑
tpma
‑
cof膜材料分别置于1m、3m、5m硝酸溶液中浸泡72小时后,对其进行红外测试。
[0045]
从红外图(图9c)中可以看出,t
‑
tpma
‑
cof膜材料的特征峰位置与强度均没有发生明显的变化,证明其具有较强的酸稳定性。
[0046]
实施例3t
‑
tpma
‑
cof膜材料的过滤实验
[0047]
制备的t
‑
tpma
‑
cof膜材料的过滤实验,是通过一个订制的u型玻璃过滤装置进行。这个过滤装置被t
‑
tpma
‑
cof膜材料分为原液和滤液两部分。进行过滤实验时,在装置的中
间处夹入一张制备的t
‑
tpma
‑
cof膜材料后,原液部分装入一定hno3(hcl)浓度下离子浓度相同的多离子溶液,滤液部分装入相同体积的水溶液以平衡渗透压,同时利用ph计监测水溶液的h
+
浓度。两部分装入溶液后离子开始从原液部分渗透,过滤实验即开始。两部分均使用电磁搅拌以促进渗透作用的进行。每隔一小时,两部分各取10ml溶液,利用icp
‑
oes测定离子浓度。复用性实验在首次过滤实验完成后,不拆开过滤装置,直接将两部分溶液倒出,再在两部分均装入1m hno3溶液,电磁搅拌清洗2天,以除去可能残留于装置内壁或膜内的离子。随后,将清洗用的1m hno3溶液倒出,再在两部分分别装入新的多离子溶液和水溶液,开始同一张膜的第二次复用实验,每进行完一次过滤实验,依上述步骤再次进行清洗。订制的u型玻璃过滤装置最大容积为1000ml,每次进行过滤实验时,原液部分与滤液部分各装入400ml溶液。
[0048]
为了测试所制备t
‑
tpma
‑
cof膜材料亲水面和疏水面对水的通透性能,仅在装置的一边的滤杯中加入水溶液,每隔30min测量滤过的水的体积,通过换算得到水的过滤速率。
[0049]
1)水的透过率
[0050]
由于t
‑
tpma
‑
cof滤膜的两个面分别具有亲水和疏水的性质,本发明分别探究了t
‑
tpma
‑
cof膜的两个面对水的渗透性能。研究发现(图5a),亲水面对水的透过率可以达到2.809l m
‑2h
‑1,而疏水面对水的透过率仅为0.061l m
‑2h
‑1。这可能是由t
‑
tpma
‑
cof的两面不同的图灵结构导致的。本发明认为,亲水面孔状的图灵结构及其超亲水的特性使得水分子更容易进入并顺利通过由该结构所搭建纵向导流通道,从而大大增加了膜的透水性;而疏水面线状的图灵结构呈平面状分布且非常密集,无法构建纵向的导流通道,因而使水分子不易透过膜的表面。
[0051]
2)阳离子透过率
[0052]
接着,本发明又分别探究了t
‑
tpma
‑
cof膜的亲水面和疏水面对金属离子的筛分能力。为了系统地评估t
‑
tpma
‑
cof的离子筛分性能,实验中选用的多离子溶液含有14种阳离子,包括h
+
、碱金属、碱土金属、过渡金属、镧系和锕系金属离子,价态包括一价、二价和三价。
[0053]
多离子及染料溶液的配制:含有(na
+
、uo
22+
、nd
3+
、gd
3+
、sm
3+
、la
3+
、ce
3+
、mn
2+
、sr
2+
、ba
2+
、ni
2+
、co
2+
、zn
2+
)13种共存离子的多离子溶液是根据文献中报道的典型核工业流出物组分配制的。配制过程中,称取一定质量的相应金属氧化物或金属含氧酸盐溶解于浓硝酸中,然后加稀释定容,使多离子溶液中各金属离子的浓度均为5.0mmol l
‑1。实验中所用到不同ph的多离子溶液,是通过移取一定体积的多离子溶液加水稀释,加hno3或naoh调节ph,再定容得到的。
[0054]
实验时,水和多离子溶液分别被加入过滤器的两侧,并通过溶液浓度随时间的变化探究膜的过滤性能。首先,本发明在过滤器的疏水面一侧加入含有1m hno3的多离子溶液,另外一侧加入同样体积的水,通过ph计监测水溶液的h
+
浓度的变化,并通过icp测试监测水中金属离子的出现和浓度变化情况。实验结果表明,13种金属离子在实验条件中均不能通过t
‑
tpma
‑
cof膜,但是h
+
则可以很容易地透过该膜。水溶液的ph由6.25降低到1.28,渗透通量为54.5mol l
‑1m2。离子筛分过程中,由于渗透压的存在,水会通过膜反渗透到含有多离子溶液的一侧,使得多离子溶液的体积增加,从而不利于其在真实的乏燃料后处理环境中应用。当含有1m hno3的多离子溶液加入到过滤器的亲水面一侧时,14种金属离子同样在
实验条件下均不能通过t
‑
tpma
‑
cof膜。然而,相比于多离子溶液在疏水面一侧时,可以明显地观察到滤液中h
+
的浓度增加速度极大地提高了,在相同时间内ph由6.40降低到0.94,渗透通量为119.3mol l
‑1m2(如图5b、6a)。这个过程中观察到含多离子溶液的一侧没有明显的体积变化,说明当多离子溶液在亲水面时,此时疏水面特有的线状图灵结构有效地阻挡了水分子通过膜的反渗透行为,起到了缓和渗透压的作用。综上所述,多离子溶液在过滤器的亲水面一侧加入时,离子筛分的效率更高,因此后续实验均将多离子溶液加入到亲水面一侧中进行实验。
[0055]
在核燃料后处理的几种化学转化中,浓硝酸作为反应物
‑
溶剂广泛被应用。因此,在液体废物最终浓缩和储存之前,脱硝是非常必要的。上述实验说明,t
‑
tpma
‑
cof膜材料具有只允许h
+
通过而不允许其他金属通过的性质,这一特性再加上t
‑
tpma
‑
cof膜材料优秀的酸稳定性为其用于高放废液的脱酸处理提供了重要的基础,从而可以有效避免传统处理方法因高酸度废液的稀释而造成的增容及处理难度的增加。乏燃料后处理过程中强酸性的苛刻条件使得大部分膜材料通常无法发挥其作用。以此,本发明首先探究了酸度大小对t
‑
tpma
‑
cof膜材料的过滤性能的影响。研究发现,在3m hno3条件下,水溶液的ph由6.20降低到0.45,渗透通量为368.8mol l
‑1m
‑2(如图6b)。更令人惊喜的是,在5m hno3(如图6c)条件下,t
‑
tpma
‑
cof膜材料仍然保持着高效的金属离子阻截能力,仍然只允许h
+
通过。水溶液的ph由6.21降低到0.14,渗透通量达到1015.7mol l
‑1m
‑2(如图6c)。上述研究结果表明,t
‑
tpma
‑
cof膜材料能够轻松适应乏燃料后处理过程中高酸度的苛刻条件,并能够有效地实现高酸度废液的脱酸处理。此外,由于t
‑
tpma
‑
cof膜在hno3体系中表现出了较好的金属离子阻截和h
+
透过性能,本发明继续探究了t
‑
tpma
‑
cof膜材料在盐酸体系中的筛分能力。从图6d、e中可知,在1m和3m hcl条件下,t
‑
tpma
‑
cof有效阻截了金属离子的通过,只允许h
+
通过,且h
+
的浓度变化速率较高,渗透通量达到108.8和299.8mol l
‑1m
‑2。同样,在5m hcl体系中,t
‑
tpma
‑
cof膜材料仍然保持着对h
+
高效透过能力,水溶液的ph由6.10降低到0.03,渗透通量达到970.1mol l
‑1m
‑2(如图6f)。
[0056]
接着,本发明探究了时间对过滤的影响,以ph为1.8的硝酸体系和5m的硝酸体系为例,将过滤时间延长至192小时。实验结果发现两个体系均能够在24小时内实现h
+
的快速渗透,24小时后的渗透速率较为缓慢。重要的是,192小时后,t
‑
tpma
‑
cof膜材料仍然保持着高效的金属离子阻截能力,只允许h
+
通过(图7a,b)。
[0057]
基于以上的实验结果,本发明提供了可能的脱酸机理。如图8所示,在过滤实验中,t
‑
tpma
‑
cof膜材料上的富电子基团(e.g.n/o)首先会与溶液中的金属阳离子发生相互作用,并使其固定在膜的孔道中。此时,处于高价态和大离子直径的金属离子更容易与这些富电子基团结合并固定下来,于是,原有的离子传输通道由于这些金属离子的固定而变得更小。当其它阳离子想要通过时,就会受到已固定的金属阳离子的电荷排斥效应,从而难以顺利通过。而氢离子由于具有最低的价态和最小的离子直径,受到的电荷排斥作用最小,因此,在浓度差驱动作用下,可轻易通过t
‑
tpma
‑
cof膜材料。对于溶液中的阴离子,固定的金属阳离子所产生的电荷斥力将不起作用。因此,阴离子也能顺利通过t
‑
tpma
‑
cof膜材料,以维持电荷平衡,这一结果已通过离子色谱分析得到证实(图7c,d)。
[0058]
本发明基于cofs(covalent organic frameworks materials,共价有机框架材料)材料优秀理化稳定性和独特的结构特性,采用界面法首次生长出双向异性图灵结构的
t
‑
tpma
‑
cof膜材料,并应用于脱酸领域。该膜材料具有明显的两面各异性,一面具有线状的图灵结构和疏水的性质,另一面具有孔状的图灵结构和超亲水的性质。独特结构使亲水面相对于疏水面对水的透过率提高了45倍,可在保证膜透水性的同时,有效抑制水的反向渗透。此外,上述膜材料具有优秀酸稳定性、热稳定性以及辐照稳定性,能够轻松适应乏燃料后处理料液高温、髙酸和强辐射的极端环境。渗透实验表明,t
‑
tpma
‑
cof膜材料复杂多离子体系中对金属离子具有极高的截留能力,在酸度高达5m的hno3模拟料液中进行超过192小时的过滤实验,金属离子阻截率为100%,而氢离子则可轻易透过该膜,渗透通量也高达1015.7mol l
‑1m
‑2,可有效实现高酸度料液的高效脱酸处理。本发明为新型图灵结构膜材料的设计和制备提供了新的思路,同时为乏燃料后处理料液等高酸度工业废水的脱酸减容和处理流程简化提供了新的解决途径。
[0059]
参考文献:
[0060]
1.a.turing,phil.trans.r.soc.lond.b,1952,237,37
‑
72。
[0061]
2.v.v.castets,e.dulos,j.boissonade and p.de kepper,phys.rev.lett.,1990,64,2953
‑
2956。
[0062]
3.b.hasslacher,r.kapral and a.lawniczak,chaos,1993,3,7
‑
13。
[0063]
4.a.p.a.john ross,j.phys.chem.,1995,99,10417
‑
10419。
[0064]
5.p.de kepper,v.castets,e.dulos and j.boissonade,physica d,1991,49,161
‑
169。
[0065]
6.w.yang,y.zhu,z.sun,c.gao and l.xue,adv.mater.interfaces,2019,6。
[0066]
7.x.luo,x.lu,y.chen,x.chen,h.guo,c.song,n.wang,d.su,g.wang,l.cui and y.liu,mater.adv.,2020,1,3449
‑
3459。
[0067]
8.z.tan,s.chen,x.peng,l.zhang and c.gao,science,2018,360,518
‑
521。
[0068]
9.k.shen,p.li,t.zhang and x.wang,j.membr.sci.,2020,607。
[0069]
10.c.jiao,x.song,x.zhang,l.sun and h.jiang,acs appl.mater.interfaces.,2021,13,18380
‑
18388。
[0070]
11.x.l.zhang,p.p.yang,y.r.zheng,y.duan,s.j.hu,t.ma,f.y.gao,z.z.niu,z.z.wu,s.qin,l.p.chi,x.yu,r.wu,c.gu,c.m.wang,x.s.zheng,x.zheng,j.f.zhu and m.r.gao,angew.chem.int.ed.engl.,2021,60,6553
‑
6560。
[0071]
12.a.k.kota,g.kwon,w.choi,j.m.mabry and a.tuteja,nat.commun.,2012,3,1025。
[0072]
13.j.r.werber,c.o.osuji and m.elimelech,nat.rev.mater.,2016,1,1
‑
15。
[0073]
14.z.yin,y.zheng,h.wang,j.li,q.zhu,y.wang,n.ma,g.hu,b.he,a.knop
‑
gericke,r.schlogl and d.ma,acs nano,2017,11,12365
‑
12377。
[0074]
15.j.zhu,l.wang,j.wang,f.wang,m.tian,s.zheng,n.shao,l.wang and m.he,acs nano,2020,14,15306
‑
15316。
[0075]
16.l.wang,x.guo,k.cao,b.li,y.li,m.zhang,r.wen,x.li,s.li and l.ma,j.mater.chem.a,2017,5,8051
‑
8061。
[0076]
17.z.wang,z.wang,s.lin,h.jin,s.gao,y.zhu and j.jin,nat.commun.,2018,9,2004。
[0077]
18.b.liang,x.he,j.hou,l.li and z.tang,adv.mater.,2019,31,e1806090。
[0078]
19.a.a.uliana,n.t.bui,j.kamcev,m.k.taylor,j.j.urban and j.r.long,science,2021,372,296
‑
299。
[0079]
20.d.d.medina,v.werner,f.auras,r.tautz,m.dogru,j.schuster,s.linke,m.doblinger,j.feldmann,p.knochel and t.bein,acs nano,2014,8,4042
‑
4052。
[0080]
21.c.cui,c.fan,y.wu,m.xiao,t.wu,d.zhang,x.chen,b.liu,z.xu,b.qu and w.liu,adv.mater.,2019,31,e1905761。
[0081]
22.w.yang,m.zhao,y.ban,k.yang,y.zhou,n.cao and y.wang,angew.chem.int.ed.engl.,2021,doi:10.1002/anie.202108185。
[0082]
23.g.zakrzewska
‑
trznadel,desalination,2013,321,119
‑
130。
[0083]
24.l.xian,g.tian,c.m.beavers,s.j.teat and d.k.shuh,angew.chem.int.ed.engl.,2016,55,4671
‑
4673。
[0084]
25.s.wang and y.li,j.electroanal.chem.,2004,571,37
‑
42。
[0085]
26.s.w.g roth,nucl.eng.des.,2000,202,197
‑
207。
[0086]
27.a.dakshinamoorthy,p.s.dhami,p.w.naik,n.l.dudwadkar,s.k.munshi,p.k.dey and v.venugopal,desalination,2008,232,26
‑
36。
[0087]
28.m.s.m.s.krehula,e.kuzmann and z.homonnay,j.radioanal.nucl.chem.,2019,322,1477
‑
1485。
[0088]
29.s.mishra,k.chandran,m.lavanya and n.k.pandey,ann.nucl.energy,2020,138。
[0089]
30.m.w.cooke,a.botti,d.zok,g.steinhauser and k.r.ungar,proc.natl.acad.sci.u.s.a.,2020,117,14703
‑
14711。
[0090]
31.m.leblanc,g.leturcq,e.welcomme,x.deschanels and t.delahaye,j.nucl.mater.,2019,519,157
‑
165。
[0091]
32.m.g.ferrier,b.w.stein,s.e.bone,s.k.cary,a.s.ditter,s.a.kozimor,j.s.lezama pacheco,v.mocko and g.t.seidler,chem sci,2018,9,7078
‑
7090。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1