一种HEK293细胞瞬时表达转染用试剂和瞬时表达系统转染方法与流程
一种hek293细胞瞬时表达转染用试剂和瞬时表达系统转染方法
技术领域
1.本发明涉及基因工程技术领域,尤其涉及一种hek293细胞瞬时表达转染用试剂和瞬时表达系统转染方法。
背景技术:
2.全球医药产业中生物药物的市场比重不断扩大,市场规模也稳步增长。其中重组蛋白质(包括抗体)药物的生产是生物制药的一个重要部分。在2018年,全球生物药物市场规模已高达2618亿美元,其中单抗药物为1448亿美元(占比高达55.3%)。细菌、酵母、昆虫细胞都可以作为重组蛋白药物表达的宿主细胞,但由于哺乳动物细胞具有类似人类细胞的翻译后加工修饰(post-translational modifications,ptms),已经成为目前重组蛋白生产的重要平台。2015~2018年新批准上市的重组抗体,79%来源于哺乳动物细胞。尽管经fda批准上市的重组蛋白质(抗体)药物大都是由cho细胞生产的,但cho细胞系中诸多的糖基化方式与人源细胞不尽相同,产生的重组蛋白仍可能有一定的免疫原性。hek293作为人源化的细胞系,表达重组蛋白药物与来源于人的蛋白具有完全一致的ptms,具有不会引起免疫反应等优点。
3.目前哺乳动物细胞生产的最耗时的方法是建立稳定的细胞系,而瞬时表达系统(transient gene expression,tge)无需进行复杂和长时间的筛选步骤,仅通过一个批次的培养就可以收获目的蛋白。瞬时表达系统高效率特点使得在快速获得流感检测抗体、筛选大量蛋白候选药物以及个体化小规模蛋白药物生产等方面具有无可替代的优势。在tge系统中,通过瞬时转染的方法将携带目的基因的质粒直接转入细胞驱动重组蛋白的生产。转染后,包含目的基因的质粒(pdna)基本不与细胞中染色体基因组dna发生整合,直接进行重组蛋白的转录和翻译过程。细胞在分批培养过程中,细胞中的质粒随着细胞的分裂而被稀释,并在连续的几轮细胞分裂中丢失。典型的表达窗口是在质粒完全丢失前,转染后的24~96h。通常7天左右就可以完成一轮瞬时表达蛋白的收获。提高tge的性能就需要从tge系统中涉及的过程入手进行优化,包括但不限于针对递送试剂、转染方式、转染条件、表达载体、培养工艺的优化和培养基等的优化。瞬时表达系统单位体积产量与稳定哺乳动物细胞株的单位体积产量差距较大,这也是大规模基因瞬时表达技术所面临的最大瓶颈。为了改善这一现状,对tge系统进行优化是目前研究的一个重点。
技术实现要素:
4.本发明的目的在于提供一种hek293细胞瞬时表达转染用试剂和瞬时表达系统转染方法,能更好的提高hek293细胞外源蛋白表达水平,高效表达目的基因。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种hek293细胞瞬时表达转染用试剂,所述试剂包括穿膜肽dlr8与脂质体混合物,试剂中穿膜肽dlr8:脂质体的质量比为4~8.8:1。
7.优选的,所述hek293细胞瞬时表达转染用试剂中,穿膜肽dlr8的氨基酸序列如seq id no.1所示,其中第1、3、5位氨基酸为d-精氨酸。
8.本发明还提供了所述hek293细胞瞬时表达转染用试剂的转染方法,包括以下步骤:
9.(1)将穿膜肽dlr8、包含目的基因的质粒、脂质体分别与孵育buffer混合,孵育3~7min;然后,将穿膜肽dlr8孵育物与脂质体孵育物混合,再孵育8~12min获得转染试剂孵育物;将所述转染试剂孵育物与包含目的基因的质粒孵育物,混合,孵育8~12min,得转染复合物;
10.(2)将所述转染复合物滴加到hek293细胞中,静置培养6~8h后更换培养基,再继续培养24~48h,完成细胞转染;
11.(3)细胞转染处理24~72h后,将细胞接种于293细胞无血清培养基,进行悬浮培养。
12.优选的,步骤(1)中采用的孵育buffer为高糖dmem溶液。
13.优选的,步骤(1)转染复合物中包含目的基因的质粒的浓度为1.2~2.0μg/ml,脂质体的浓度为3~5μl/ml。
14.优选的,步骤(1)转染复合物中,穿膜肽dlr8与包含目的基因的质粒的质量比为1:2~4。
15.优选的,步骤(2)中滴加的转染复合物用量与细胞密度的比例为1.25~5ml:1~4
×
105。
16.优选的,步骤(2)中更换所用培养基为高糖dmem完全培养基。
17.优选的,步骤(2)中静置培养的温度为37℃,co2的体积分数为3~7%。
18.优选的,步骤(3)进行悬浮培养过程中,加入1~3mm的丁酸钠。
19.本发明的技术效果和优点:
20.本发明提供了一种hek293细胞瞬时表达转染用试剂和瞬时表达系统转染方法,本发明中选择穿膜肽dlr8与脂质体混合物作为hek293细胞瞬时表达的转染试剂,高糖dmem培养基作为转染孵育buffer,并在细胞转染处理后48h进行细胞悬浮,添加小分子添加剂丁酸钠,提高了hek293细胞外源蛋白的表达水平,使得目的基因可高效表达。
附图说明
21.图1为多肽凝胶阻滞电泳图;
22.图2为cpp-lip-pdna模式介导的基因递送图(a:不同剂量dlr8使用下dlr8-lip-pdna模式介导的egfp表达48h荧光图;b:不同剂量cppmk2使用下cppmk2-lip-pdna模式介导的egfp表达48h荧光图;c:对图2adlr8-lip-pdna转染效率统计分析的结果;d:对图2b cppmk2-lip-pdna转染效率统计分析的结果);
23.图3为dlr8-lip-pdna模式介导的基因递送图(a:dlr8-lip-pdna模式在更大dlr8剂量范围内的测试结果;b:dlr8-lip-pdna在最佳工作范围内对细胞无毒性;c:凝胶阻滞验证dlr8溶液在4℃保存3个月后对dna的阻滞能力);
24.图4为不同转染孵育buffer转染效率对比(a:实验组egfp的荧光图b:转染效率统计分析柱形图);
25.图5为不同悬浮培养基性能测试(a:不同悬浮培养基分批培养seap的体积产量;b:不同悬浮培养基活细胞密度与细胞活率);
26.图6为不同pdna(包含目的基因的质粒)浓度转染seap表达量;
27.图7为转染后不同时间悬浮egfp、seap表达水平;(a:不同悬浮时间48h egfp荧光图;b:不同悬浮培养基活细胞密度与细胞活率);
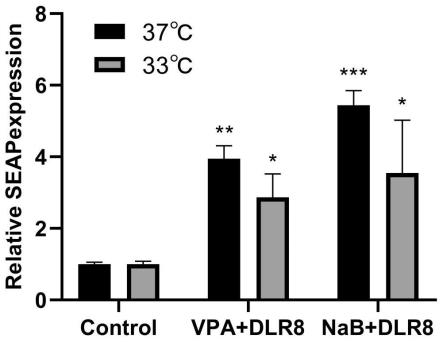
28.图8为实验例3所述各实验组对seap表达量的影响。
具体实施方式
29.本发明提供了一种hek293细胞瞬时表达转染用试剂,所述试剂包括穿膜肽dlr8与脂质体混合物,试剂中穿膜肽dlr8:脂质体的质量比为4~8.8:1。
30.在本发明所述转染用试剂中,所述穿膜肽dlr8:脂质体的质量比优选为4~8.8:1,进一步优选为5~7:1,再进一步优选为6:1。
31.在本发明所述转染用试剂中,所述穿膜肽dlr8的氨基酸序列优选为如seq id no.1所示,其中第1、3、5位氨基酸为d-精氨酸;d-精氨酸可以提高多肽的抗降解能力,提高其稳定性,1/3/5设置为d-精氨酸,既保证了穿膜活性又提高了多肽的稳定性,是最优的选择;单独的穿膜肽dlr8无法直接介导质粒dna的高效表达,穿膜肽dlr8与和脂质体组合使用后可以发挥协同作用,实现更高的转染效率和蛋白表达。
32.本发明还提供了所述hek293细胞瞬时表达转染用试剂的转染方法,包括以下步骤:
33.(1)将穿膜肽dlr8、包含目的基因的质粒、脂质体分别与孵育buffer混合,孵育3~7min;然后,将穿膜肽dlr8孵育物与脂质体孵育物混合,再孵育8~12min获得转染试剂孵育物;将所述转染试剂孵育物与包含目的基因的质粒孵育物,混合,孵育8~12min,得转染复合物;
34.(2)将所述转染复合物滴加到hek293细胞中,静置培养6~8h后更换培养基,再继续培养24~48h,完成细胞转染;
35.(3)细胞转染处理24~48h后,将细胞接种于293细胞无血清培养基,进行悬浮培养。
36.在本发明所述转染方法步骤(1)中,采用的孵育buffer优选为高糖dmem溶液,使用高糖dmem溶液作为孵育buffer获得的转染效率最高。
37.在本发明所述转染方法步骤(1)中,穿膜肽dlr8、包含目的基因的质粒、脂质体分别与孵育buffer混合的孵育时间优选为3~7min,进一步优选为4~6min,再进一步优选为4.5~5.5min。
38.在本发明所述转染方法步骤(1)中,将穿膜肽dlr8孵育物与脂质体孵育物混合再孵育的时间优选为8~12min,进一步优选为9~11min,再进一步优选为9.5~10.5min。
39.在本发明所述转染方法步骤(1)中,穿膜肽dlr8孵育物与脂质体孵育物混合孵育完成后,再加入包含目的基因的质粒孵育物,混匀,最后进行孵育,得转染复合物,孵育时间优选为8~12min,进一步优选为9~11min,再进一步优选为10min。
40.在本发明所述转染方法步骤(1)转染复合物中,包含目的基因的质粒的浓度优选为1.2~2.0μg/ml,进一步优选为1.4~1.8μg/ml,再进一步优选为1.6μg/ml;脂质体的浓度
优选为3~5μg/ml,进一步优选为3.5~4.5μg/ml,再进一步优选为4μg/ml。
41.在本发明所述转染方法步骤(1)转染复合物中,穿膜肽dlr8与包含目的基因的质粒的质量比优选为1:2~4,进一步优选为1:2.2~3.8,再进一步优选为1:2.4~3.6。
42.在本发明所述转染方法步骤(2)中,将转染复合物滴加到hek293细胞中,滴加的转染复合物用量与细胞密度的比例优选为1.25~5ml:1~4
×
105,进一步优选为1.75~4.5ml:1.5~3.5
×
105,再进一步优选为2.25~4ml:2~3
×
105。
43.在本发明所述转染方法步骤(2)中,将转染复合物滴加到hek293细胞中,静置培养更换培养基,再继续培养完成细胞转染;静置培养时间优选为6~8h,进一步优选为6.5~7.5h,再进一步优选为7h,静置培养温度优选为37℃,静置培养环境中co2的体积分数优选为3~7%,进一步优选为4~6%,再进一步优选为4.5~5.5%;更换的培养基优选为高糖dmem完全培养基;高糖dmem完全培养基为添加了10%血清的高糖dmem溶液;继续培养时间优选为24~72h,进一步优选为36~60h,再进一步优选为48h。
44.在本发明所述转染方法步骤(3)中,细胞转染处理24~48h后,将细胞接种于293细胞无血清培养基,进行悬浮培养;所述细胞悬浮时间优选为细胞转染处理24~48h后,进一步优选为细胞转染处里30~42h后,再进一步优选为36h后;在悬浮培养过程中,优选的加入丁酸钠,所述丁酸钠的浓度优选为1~3mm,进一步优选为1.5~2.5mm,再进一步优选为1.8~2.2mm;所述丁酸钠的加入时机优选为转染后6h和/或转染后48h进行悬浮培养的初期添加,进一步优选为,转染后48h进行悬浮培养时添加蛋白产量最高;丁酸钠为组蛋白去乙酰化酶抑制剂,其起到提高蛋白表达的作用。
45.在本发明所述转染方法中,所述hek293细胞系,优选的包括293s、293-h、293-f和293-t细胞系中的一种或多种。
46.下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
47.实施例1
48.本发明实施例中所使用的大肠杆菌(escherichia coli)jm109(购自生工生物工程股份有限公司)、所使用的细胞系(购自美国invitrogen公司)、试剂、工具酶均为市售商品。所用的载体为pegfp-c1载体含有增强绿色荧光蛋白(enhanced green fluorescentprotein,egfp)基因(购自美国clontech公司)。所用转染试剂pei为pei max(购自美国polysciences公司),所用转染试剂lip为lip2000(购自中国兰杰柯科技有限公司)。
49.1、细胞培养
50.hek293细胞在10%胎牛血清、dmem高糖培养基中,37℃,体积分数为5%co2条件下贴壁培养,待细胞汇合度为80~90%时,使用胰酶消化细胞,对细胞进行传代处理。新复苏的细胞至少传代3次后再进行铺板和转染。选择生长状态良好,细胞汇合度在60~90%的hek293细胞接种到24孔培养板上,待铺板密度达到约70%~90%进行转染。
51.2、穿膜肽的合成及多肽液的制备
52.多肽委托浙江宏拓生物科技有限公司采用固相合成法制备,多肽序列和分子量见表1。所有多肽均经过hplc纯化和分析(纯度≥95%),多肽的分子量通过maldi-tof-ms质谱确认。
53.表1多肽序列和分子量
[0054][0055][0056]
多肽储存溶液的制备:多肽合成后以干粉形式每管2mg分装到1.5ml ep管中,收到装有多肽干粉的ep管后,将多余的干粉放置于-80℃冻存;不要打开装干粉的ep管的盖子,将需要使用的干粉带ep管12000r/min,离心3min。在超净工作台里使用移液枪在含有2mg多肽的ep管加入500μl无菌去离子水,摇匀使多肽完全溶解配制成4mg/ml的多肽溶液,4℃储存备用。
[0057]
3、pseap-c1载体的构建
[0058]
将pegfp-c1载体上的egfp基因替换为分泌型碱性磷酸酶(secreted alkaline phosphatase,seap)基因,人工合成seap基因(如genbank:km403567.1,272—1831所示),具体交由通用生物基因(安徽)有限公司完成。为便于克隆及保证序列完整性,合成seap基因时,5’端引入aacgctagc序列,其中aac为保护碱基,gctagc为nhei酶切位点;3’端引入agatctgct序列,其中gct为保护碱基,agatct为bglii酶切位点。
[0059]
用nhei/bglii双酶切合成的seap基因,同时用nhei/bglii双酶切pegfp-c1质粒dna,再用neb公司
tm
的连接试剂盒25℃连接5min。将连接产物加入到e.coli jm109菌株感受态细胞悬液中转化,取150μl转化菌液接种到含有氨苄青霉素的lb平板上,37℃培养过夜,挑取单菌落继代培养。提取重组质粒并进行双酶切(nhei/bglii)验证,取酶切验证正确的质粒进行测序验证,构建正确的质粒命名为pseap-c1。
[0060]
4、细胞转染
[0061]
分别将dlr8、pseap-c1、脂质体在三个ep管中分别与50μl高糖dmem进行混匀孵育5min;将dlr8孵育物与脂质体孵育物混合后孵育10min。然后加入pseap-c1孵育物轻轻混匀,孵育10min形成最终的转染复合物。转染复合物中pseap-c1的浓度为1.6μg/ml,脂质体
的浓度为4μg/ml,dlr8与pseap-c1的质量比为1:3。
[0062]
在24孔板中转染完成egfp基因的转染测定,含有新鲜完全培养基的24孔板中,培养基体积为500μl,转染复合物滴入含有新鲜完全培养基的24孔板中,转染复合物用量与细胞密度的比例为3ml:2.5
×
105;转染6h后,更换新的高糖dmem完全培养基,再继续培养36h,完成细胞转染。
[0063]
5、细胞悬浮
[0064]
细胞转染处理48h后,将细胞以1.5
×
106cells/ml的密度接种于293细胞无血清培养基,添加浓度为2mm的丁酸钠进行悬浮培养,在悬浮后第4天、第6天各收集200μl细胞上清检测分泌到胞外seap的表达水平。荧光显微观察结果,如图7-a和图7-b所示。
[0065]
实验例1
[0066]
1、不同孵育buffer优化
[0067]
将pseap-c1载体转染到hek293f细胞中,分别用dmem/f12、高糖dmem、pbs三种溶液作为孵育buffer对pdna(包含目的基因的质粒)、lip2000进行孵育,转染48h后,通过倒置荧光显微镜评估不同孵育buffer的转染效率。结果如图4所示,使用高糖dmem作为孵育buffer获得的转染效率最高,pbs作为孵育buffer获得的转染效率最低。
[0068]
2、不同悬浮培养基表达性能测试
[0069]
将携带seap基因的表达载体pseap-c1转染hek293f细胞,48h后分别使用4种不同的悬浮培养基以1.5
×
106cells/ml的密度接种细胞进行分批培养。期间每天记录细胞的活性和数量,细胞活率小于60%时收获细胞上清液,使用碱性磷酸酶检测试剂盒和酶标仪对seap表达水平进行检测。4种不同的悬浮培养基为opm-293cd05 medium(上海奥浦迈生物科技股份有限公司)、freestyle
tm
293表达培养基(美国thermofisher公司)、expi293
tm
表达培养基(美国thermofisher公司)、293细胞无血清培养基(河南普诺易生物制品研究院有限公司)。其中freestyle
tm
293表达培养基、expi293
tm
表达培养基为国外知名品牌的进口高端培养基,opm-293cd05medium、293细胞无血清培养基为国内具备自主知识产权的高端无血清培养基。
[0070]
seap的检测结果表明,如图5a所示,293实验组的seap表达水平最高。对细胞的生长情况检测表明,如图5b所示,293细胞无血清培养基的生长密度偏低,freestyle
tm
293表达培养基的生长密度最高但高密度维持时间不如opm-293cd05培养基。
[0071]
3、不同pdna、lip2000(脂质体)浓度优化
[0072]
将pdna的浓度设置为1μg/ml、1.6μg/ml、2μg/ml、2.5μg/ml,pdna:lip2000(质量/体积)的比例1:2.5。孵育buffer使用高糖dmem基础培养基,悬浮培养基使用293细胞无血清培养基。
[0073]
结果如图6所示,当pdna浓度为最低浓度1μg/ml和最高浓度2.5μg/ml时,seap的表达水平较低,1.6μg/ml和2μg/ml的dna浓度下seap的表达水平较高且相当。以1μg/ml的pdna浓度为对照组,1.6μg/ml的实验组表达量有显著提升(p《0.001)。因此pdna浓度为1.6μg/ml,lip2000浓度为4μl/ml是在6孔板中转染最经济高效的使用剂量。
[0074]
4、转染后悬浮时间优化
[0075]
使用高糖dmem作为转染孵育buffer,293细胞无血清培养基作为悬浮后的表
达培养基,在6孔板中以2ml转染体系进行转染,pdna浓度设置为1.6μg/ml,lip2000浓度设置为4μl/ml,分别设置转染后6h悬浮组,12h悬浮组,24h悬浮组,48h悬浮组。转染后6h更换新鲜的高糖dmem完全培养基,在以上所述6h、12h、24h、48h时间点消化细胞,将培养方式改为悬浮分批培养。在转染后48h检测胞内egfp的表达水平,在悬浮后第4天、第6天各收集200μl细胞上清检测分泌到胞外seap的表达水平。
[0076]
荧光显微观察结果如图7-a和图7-b所示,在设置的6h~48h区间内转染后悬浮的时间越晚,egfp的表达效率越高,这与更长的培养时间下的seap表达量的检测结果趋势是一致的。但转染后悬浮时间在48h后再进行延长是不适宜的,因为在转染后48h,细胞的汇合度会大于100%,再往后推迟悬浮培养的时间可能会导致细胞发生严重的接触抑制,细胞无法贴壁和引起细胞死亡等现象。因此贴壁转染后最佳的悬浮时间在48h左右。
[0077]
实验例2
[0078]
1、凝胶阻滞实验
[0079]
带正电荷的阳离子多肽与带负电荷的pdna可以相互作用,形成稳定的cpp-pdna复合物,凝胶阻滞实验可以灵敏的检测到这种变化。将1μg pdna与不同体积的肽混合,加入高糖dmem基础培养基,使最终体积达到9μl。具体而言,分别取cppmk2储存溶液0.5μl、1μl、1.5μl、2μl、2.5μl、3μl、3.5μl、4μl,chr4储存溶液0.5μl、1μl、2μl、3μl、5μl、7μl,dlr8溶液0.25μl、0.5μl、0.75μl、1μl、1.25μl、1.5μl,在分别在上述多肽溶液中加入高糖dmem基础培养基8.5μl、8μl、7.5μl、7μl、6.5μl、6μl、5.5μl、5μl、8.5μl、8μl、7μl、6μl、4μl、2μl、8.75μl、8.5μl、8.25μl、8μl、7.75μl、7.5μl。在37℃孵育10min后,加入1μl 10
×
dna loading buffer。制备含有superred核酸染料的0.8%琼脂糖凝胶。使用电泳分离pdna和多肽复合物。使用荧光成像系统观察pdna迁移情况。实验结果如图1所示,三种多肽与pdna的质量比在4以下的低质量比下表现出对pdna良好的阻滞能力。这种优异的阻断能力表明这些cpp可以在体外与质粒载体有效结合。
[0080]
2、不同转染方法测试
[0081]
在hek293f细胞中,通过显微荧光成像评估不同转染复合物在tge状态下的egfp表达。通过在24孔板中转染完成egfp基因的转染测定。将cpp(多肽序列)-pdna-lip和cpp-lip-pdna2种不同转染模式下的转染复合物滴入含有新鲜完全培养基的24孔板中,培养基体积为500μl。转染6h后,将培养基更换为新鲜的完全培养基。cpp-pdna-lip模式与cpp-lip-pdna模式的差异在于孵育过程的不同。
[0082]
以转染24孔板为例,cpp-pdna-lip模式与cpp-lip-pdna模式的孵育过程如下:
①
cpp-pdna-lip模式:分别将不同体积的cpp、pdna、lip在三个ep管中分别与50μl高糖dmem进行混匀孵育5min。将cpp孵育物与pdna孵育物混合后孵育10min。将步骤的孵育物与lip轻轻混匀,孵育10min形成最终的转染复合物。
②
cpp-pei/lip-pdna模式:分别将不同体积的cpp、pdna、lip在三个ep管中分别与50μl高糖dmem进行混匀孵育5min。将cpp孵育物与lip孵育物混合后孵育10min。将步骤(2)的孵育物与pdna轻轻混匀,孵育10min形成最终的转染复合物。
[0083]
结果如图2-a和图2-b所示,在cpp-lip-pdna模式下,dlr8和cppmk2可以在低浓度下提高转基因表达。在dlr8浓度为4~8.8m/m和cppmk2浓度为2~8.8m/m时,转染效率显著高于对照组。cppmk2-lip-pdna递送系统的转基因表达水平整体不如dlr8-lip-pdna递送系
统。
[0084]
3、细胞毒性实验
[0085]
细胞毒性实验采用calcein-am/pi双染法,hek293f细胞以5000个细胞/孔接种在96孔板中,培养板在培养箱中预培养24h。当细胞汇合度达到约70~90%时,向培养板中加入不同体积的cpp以达到设定浓度。同时,对照组加入lip20000.5μl和超纯水(5μl)。将板孵育12h,弃去培养基。细胞用无菌pbs洗涤一次,每孔加入100μl calcein-am/pi检测工作液,37℃避光孵育30min。孵育后,使用荧光显微镜观察染色效果。
[0086]
在更大的dlr8浓度范围内研究其对转染的影响,图3-a表明dlr8在广泛的使用范围内可以实现良好的基因传递和表达效果。其中,最好的结果是在dlr8与质粒dna的质量比为2(m/m=2)的较低剂量下获得的。图3-b显示了以dlr8-lip-pdna模式转染后24h的细胞毒性。dlr8的剂量范围从0到最佳浓度。结果表明,dlr8-lip-pdna模式在最佳m/m下没有增加细胞毒性(p》0.05)。以上数据表明dlr8具有良好的稳定性和更宽的应用范围,最佳的使用剂量为m/m在2~4。
[0087]
实验例3
[0088]
使用高糖dmem作为转染孵育buffer,293细胞无血清培养基作为悬浮后的表达培养基,在6孔板中以2ml的dlr8-lip-pdna转染体系进行转染,dna浓度设置为1.6μg/ml,lip2000浓度设置为4μl/ml,转染后6h、24h更换新鲜的高糖dmem完全培养基,转染后48h消化细胞,使用293细胞无血清培养基悬浮细胞,放置于37℃、5%co2、110r/min摇床上进行一个批次的分批培养,本方法为方案a。
[0089]
使用小分子添加剂丁酸钠(nab)或丙戊酸钠(vpa)和低温培养优化工艺。实验分为6组:
[0090]
(1)对照组:方案a;
[0091]
(2)实验组:方案a中悬浮细胞为33℃低温培养,其他条件不变;
[0092]
(3)实验组:方案a中悬浮细胞为37℃培养+4mm vpa+dlr8;
[0093]
(4)实验组:方案a中悬浮细胞为33℃培养+4mm vpa+dlr8;
[0094]
(5)实验组:方案a中悬浮细胞为37℃培养+2mm nab+dlr8;
[0095]
(6)实验组:方案a中悬浮细胞为33℃培养+2mm nab+dlr8。
[0096]
结果如图8所示,2mm nab+dlr8实验组表达量最高,与方案a相比提高了2.18倍(p《0.001),4mm vpa+dlr8实验组提高了1.58倍(p《0.05)。33℃培养下的所有实验组表达量与方案a无显著差异,低温显著抑制了两个有效提高实验组的表达水平。
[0097]
由以上实施例可知,本发明提供了一种hek293细胞瞬时表达转染用试剂和瞬时表达系统转染方法,能更好的提高hek293细胞外源蛋白表达水平,高效表达目的基因。
[0098]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1