一类A3B型不对称金属铂卟啉有机发光材料及其应用的制作方法
本发明属于光电材料技术领域,具体涉及一类a3b型不对称金属铂卟啉有机发光材料及其应用。
背景技术:
近年来,有机电致发光(oled)材料已经取得了较大进展,针对oled显示器的三原色(红、绿、蓝)都已问世,其发光材料不管在亮度、效率上也基本都能够满足要求。但是,oled显示器中有机发光材料仍然存在一些问题,比如:红光材料颜色饱和度不够,近红外发光材料还十分稀有等严重影响着显示器的发展。当前已发展的oled红光材料基本都含有吸电子基团(受体)与给电子基团(给体),然而,这些基团之间存在比较强的静电作用与π-π相互作用,这些作用造成红光oled材料的电压稳定性差。为了降低红光材料之间的相互作用,一般采用掺杂的方法,稀释红光材料的浓度。因此制备出对电压稳定的红光发射材料,对oled的发展具有重大意义。
a3b型不对称金属铂卟啉是重要的红光及近红外磷光发光材料分子,由于a3b不对称卟啉上引入的重金属铂,使其和有机配体之间的形成强的自旋偶合作用,导致处于激发单线态的电子可以有效地进行系间蹿越而回到激发三线态,同时可以使处于激发三线态的分子可以进行高效地辐射转变释放能量回到基态,从而室温下即可高效地发出磷光,并且理论磷光量子效率可以高达100%;从而使这类金属有机配合物可以作为有机电致磷光发光材料而在近红外显示和检测方面有着广泛的应用前景。
k.m.smith等对卟啉配体的进行修饰,发展了一系列合成新型卟啉配体的方法,这为合成金属铂卟啉奠定了基础,有利于进一步改进铂卟啉的发光性质(jphyschemb,2001,105,6396)。1998年forrest课题组报道了采用对称型铂金属卟啉ptoep(式1a)作为主体材料的掺杂材料,获得了红色发光材料,且最高内外量子效率分别达到了4%和23%(nat.,1998,395,151)。c.n.zhu等人进一步研究研究发现通过引入氟原子有利于降低氧化过中材料的衰退,提高铂卟啉的稳定性(inorgchem,2004,43,3724)。c.m.che等合成了高效稳定的ptf20tpp磷光材料(式1b),可以很好的将其用作红光oled中的掺杂材料(j.materchem,2003,13,1362)。
在发光材料方面,先前报道的一些金属铂卟啉均为对称型铂金属卟啉配合物,而对于a3b不对称型金属铂卟啉配合物发光材料却鲜有报道。相对于对称型铂卟啉来说,通过分别调节卟啉5,10,15,20位上(如式2)的基团(给电子或吸电子集团)来调节mlct(pt→ligand),从而使a3b型不对称铂卟啉的光物理性质更加易于调节,发展一系列化学性质稳定、结构和最低激发三线态能量易于调节红光及近红外磷光发光材料。
a3b型卟啉配体的分子设计有助于调节其π*轨道和金属d轨道的相对能量,使a3b型不对称铂卟啉的发光颜色和光谱波峰宽度和形状更加易于调节(highlyefficientoledswithphosphorescentmaterials,wileyvch,2008)。同时,铂金属卟啉配合物为四齿型中性分子,具有很好的稳定性和刚性,有利于进一步在器件中的应用。因此,a3b型不对称铂卟啉类配合物的合成发展,可以极大地促进oled等相关领域的发展。
综上所述,a3b型不对称铂卟啉配合物发光材料的合成发展会对整个红光及近红外发光材料的发展起到巨大的推动作用。
技术实现要素:
本发明的目的之一在于提供一种新型a3b型不对称金属铂卟啉配合物发光材料。
为了达到上述目的,本发明采用的技术方案为:
一种如式i所示的a3b型不对称金属铂卟啉配合物发光材料:
式i中,
r1或r2各自独立为氢、卤素、c1-c20直链烷基、甲氧基、氰基、羧基或硝基;其中,所述的r1与r2不同时为同一取代基。
进一步,优选所述的发光材料为下列之一:
再进一步,推荐本发明所述的发光材料具体按照如下步骤进行制备:
(1)以式ⅱ所示的醛a和式ⅲ所示的醛b为原料,加入吡咯,在混酸溶液中,在氮气保护下,以三氟乙酸为催化剂,醋酸酐为除水剂,在回流温度下反应完全,通常在100~130℃下回流反应2小时,得到反应混合液a;所述的混酸溶液为体积比为1:0~1的醋酸和丙酸的混合溶液;所述的式ⅱ所示的醛a、式ⅲ所示的醛b、吡咯、三氟乙酸、醋酸酐的物质的量之比为3:1:4:0.1~0.4:2.2~16(优选为3:1:4:0.2:4.2);所述的混酸溶液的加入总量以式ⅲ所示的醛b的物质的量计为16ml/mmol;
(2)向步骤(1)所得反应混合液a中加入氧化剂硝基苯,在回流温度下反应完全,通常在100~130℃下回流反应2小时,反应结束后,待反应体系冷却至常温,向所得反应混合液b中加入甲醇溶液,在0℃下静置12小时,抽滤,将所得滤渣经甲醇洗涤、干燥,得到式i所示的a3b型不对称卟啉类化合物;所述的氧化剂的加入量以式ⅲ所示的醛b的物质的量计为4.0~40mmol/mmol(优选为8mmol/mmol);
(3)将步骤(2)所得a3b型不对称卟啉化合物和二氯化铂溶于有机溶剂苯甲腈中,在回流温度下反应完全,通常在190~200℃下回流8小时,反应结束后,所得反应液c经硅胶柱色谱分离纯化,以体积比为1:0~3:1的石油醚/二氯甲烷的混合溶剂为洗脱剂进行洗脱,收集含目标产物的洗脱液并减压蒸除溶剂得到a3b型不对称铂金属卟啉类化合物;所述的a3b型不对称卟啉化合物与二氯化铂的物质的量之比为1:3~4;所述的苯甲腈的加入量以所述的a3b型不对称卟啉化合物的物质的量计为700ml/mmola3b。
式ⅱ或式ⅲ中,
r1或r2各自独立为氢、卤素、c1-c20直链烷基、甲氧基、氰基、羧基或硝基;其中,所述的r1与r2不同时为同一取代基。
本发明所述的“醛a”“醛b”没有特殊的含义,标记为“a”、“b”只是用于区分不同的物质。
本发明的目的之二在于将所述的发光材料作为红光掺杂材料用于制备oled器件。
与现有技术相比,本发明的有益效果主要在于:
(1)针对a3b型不对称铂卟啉,基团r1不等于r2,可以通过调节r1,r2,加强电荷转移吸收与荧光发射强度,从而提高发光效率。
(2)金属铂卟啉是发射光谱半峰宽很小,颜色纯度高,适用于制备红光和近红外发光材料。
附图说明
图1为实施例5中a3b不对称型铂金属卟啉配合物05的发射光谱图。
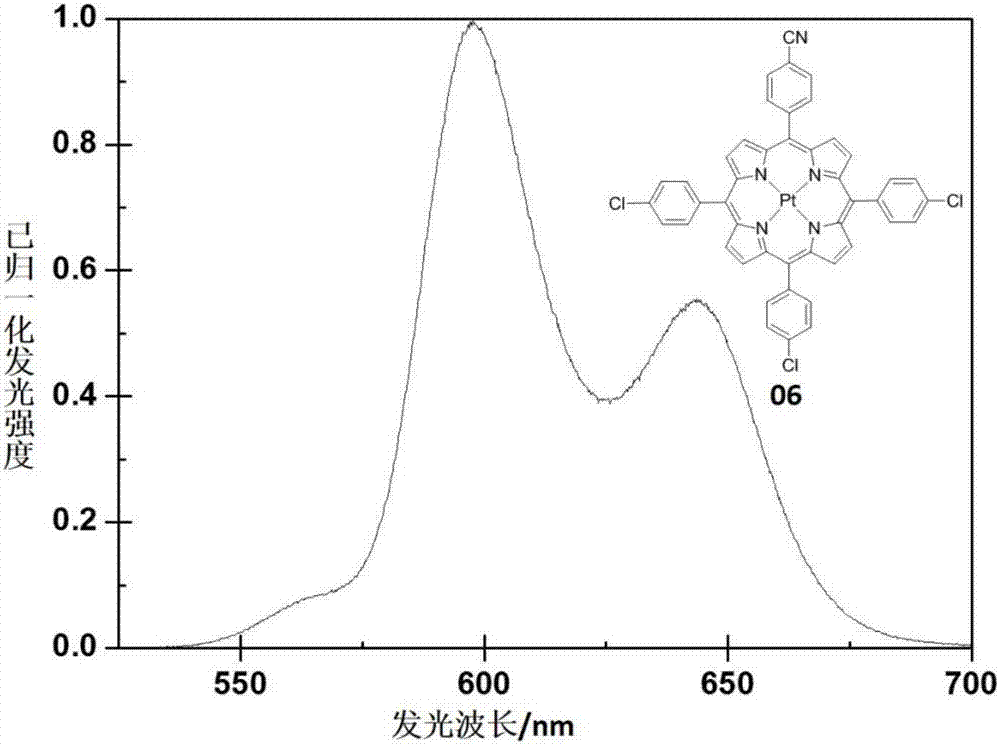
图2为实施例6中a3b不对称型铂金属卟啉配合物06的发射光谱图。
图3为实施例8中a3b不对称型铂金属卟啉配合物08的发射光谱图。
图4为实施例9中a3b不对称型铂金属卟啉配合物09的发射光谱图。
图5为实施例10中a3b不对称型铂金属卟啉配合物10的发射光谱图。
图6为实施例11中a3b不对称型铂金属卟啉配合物11的发射光谱图。
具体实施方式
实施例1:a3b不对称型铂金属卟啉配合物01的合成
配体1的合成:向带有磁力转子的干燥100ml三口烧瓶中加入对氯苯甲醛(1.05g,7.5mmol,3.0eq)。抽换氮气三次,然后加入对甲氧基苯甲醛(304ul,2.5mmol,1.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯,继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉381.4mg,取50mg通过硅胶柱色谱分离纯化,湿法上样,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体37mg,收率15%。1hnmr(500mhz,cdcl3):δ-2.81(s,2h),4.12(s,3h),7.32(d,j=8.5hz,2h),7.76(d,j=8.5hz,6h),8.12-8.16(m,8h),8.84(d,j=8.0hz,6h),8.93(d,j=5.0hz,2h).
a3b不对称型铂金属卟啉配合物01的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体1(34.4mg,0.046mmol,1.0eq),二氯化铂(37.0mg,0.14mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=4:1-1:1),得目标产物红色固体27.9mg,收率64%。1hnmr(500mhz,cdcl3):δ4.11(s,3h),7.30(dd,j=7.0,2.0hz,2h),7.73(td,j=8.5,2.5hz,6h),8.12(dd,j=6.5,2.0hz,2h),8.13-8.15(m,6h),8.92-8.94(m,6h),9.01(d,j=5.0hz,2h)
实施例2:a3b不对称型铂金属卟啉配合物02的合成
配体2的合成:向干燥100ml三口烧瓶加入磁子,抽换氮气三次。然后加入对氟苯甲醛(268ul,2.5mmol,1.0eq),对甲基苯甲醛(888ul,7.5mmol,3.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉276mg,取43mg通过硅胶柱色谱分离纯化,湿法上样,淋洗剂(石油醚/二氯甲烷=5:1-2:1),得目标产物紫色固体41mg,收率15%。1hnmr(500mhz,cdcl3):δ-2.79(t,j=8.0hz,2h),2.70(s,9h),7.43-7.49(m,2h),7.56(dd,j=8.0,2.0hz,6h),8.10(d,j=7.5hz,6h),8.15-8.18(m,2h),8.76-8.82(m,2h),8.84-8.92(m,6h).
a3b不对称型铂金属卟啉配合物02的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体2(31mg,0.046mmol,1.0eq),二氯化铂(37.0mg,0.14mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=4:1-1:1),得目标产物红色固体19mg,收率48%。1hnmr(500mhz,cdcl3):δ2.73(s,9h),7.44-7.49(m,2h),7.57(dd,j=8.0,2.0hz,6h),8.12(d,j=7.5hz,6h),8.19(td,j=5.5,2.5hz,6h),8.91-8.94(m,2h),8.97-9.01(m,6h).
实施例3:a3b不对称型铂金属卟啉配合物03的合成
配体3的合成:向干燥100ml三口烧瓶加入磁子,抽换氮气三次。然后加入对甲氧基苯甲醛(304ul,2.5mmol,1.0eq),对乙基苯甲醛(1.03ml,7.5mmol,3.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉185mg,取47mg通过硅胶柱色谱分离纯化,湿法上样,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体24mg,收率5%。1hnmr(500mhz,cdcl3):δ-2.76(s,2h),1.54(t,j=7.5hz,9h),3.01(q,j=7.5hz,6h),4.10(s,3h),7.29(dd,j=6.5,2.0hz,2h),7.58(d,j=8.0hz,6h),8.12(d,j=8.0hz,6h),8.13(d,j=9.0hz,2h),8.86(s,8h).
a3b不对称型铂金属卟啉配合物03的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体3(34mg,0.046mmol,1.0eq),二氯化铂(37.0mg,0.14mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=4:1-1:1),得目标产物红色固体29mg,收率68%。1hnmr(500mhz,cdcl3):δ1.55(t,j=3.0hz,9h),3.02(q,j=7.5hz,6h),4.11(s,3h),7.29(dd,j=6.5,2.0hz,2h),8.12-8.14(m,8h),8.96-8.98(m,8h).
实施例4:a3b不对称型铂金属卟啉配合物04的合成
配体4的合成:向带有磁子的干燥100ml三口烧瓶中加入对羧基苯甲醛(375mg,2.5mmol,1.0eq),对氯苯甲醛(1.05g,7.5mmol,3.0eq),抽换氮气三次。随后加入三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉315mg,取251mg通过硅胶柱色谱分离纯化,湿法上样,淋洗剂(石油醚/甲醇=100:0.5-100:1),得目标产物紫色固体27mg,收率2%。1hnmr(500mhz,cdcl3):δ-2.85(s,2h),7.76(d,j=8.5hz,6h),8.02(br,1h),8.14(d,j=8.0hz,6h),8.33(d,j=8.5hz,2h),8.50(d,j=8.0hz,2h),8.82-8.83(m,2h),8.85(s,6h).
a3b不对称型铂金属卟啉配合物04的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体4(27mg,0.035mmol,1.0eq),二氯化铂(29.0mg,0.11mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(二氯甲烷:甲醇=1:0-100:7),得目标产物红色固体10mg,收率30%。1hnmr(500mhz,cdcl3):δ7.77(d,j=8.0hz,6h),8.09(d,j=8.0hz,2h),8.16(d,j=8.5hz,6h),8.36(d,j=8.5hz,2h),8.94(d,j=4.5hz,2h),8.97(d,j=4.0hz,6h)。
实施例5:a3b不对称型铂金属卟啉配合物05的合成
配体5的合成:向带有磁子的干燥100ml三口烧瓶中加入对羧基苯甲醛(1.39g,7.5mmol,3.0eq),抽换氮气三次。加入对甲氧基苯甲醛(304ul,7.5mmol,3.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉941mg,取308mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体59mg,收率8%。1hnmr(500mhz,cdcl3):δ-2.84(s,2h),4.11(s,3h),7.30(d,j=8.5hz,2h),7.90(d,j=8.0hz,6h),7.08(dd,j=8.5,2.0hz,6h),8.12(dd,j=6.5,2.0hz,2h),8.82-8.83(m,6h),8.91(d,j=5.0hz,2h).
a3b不对称型铂金属卟啉配合物05的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体5(59mg,0.067mmol,1.0eq),二氯化铂(53mg,0.20mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(50ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=4:1–1:1),得目标产物红色固体17mg,收率23%。1hnmr(500mhz,cdcl3):δ4.13(s,3h),7.32(dd,j=6.5,2.0hz,2h),7.92(d,j=8.5hz,6h),8.10(dd,j=6.5,1.0hz,6h),8.14(dd,j=6.5,1.5hz,2h),8.94-8.96(m,6h),9.03(d,j=4.5hz,6h).其发射光谱图见图1.
实施例6:a3b不对称型铂金属卟啉配合物06的合成
配体6的合成:向带有磁子的干燥100ml三口烧瓶中加入对氰基苯甲醛(328g,2.5mmol,1.0eq),对氯苯甲醛(1.05g,7.5mmol,3.0eq)抽换氮气三次。随后加入三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉614mg,取401mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体209mg,收率11%。1hnmr(500mhz,cdcl3):δ-2.87(s,2h),7.76(d,j=8.5hz,6h),8.08(d,j=8.5hz,2h),8.13(d,j=8.5hz,6h),8.33(d,j=8.0hz,2h),8.75(d,j=4.5hz,2h),8.86(d,j=9.0hz,6h).
a3b不对称型铂金属卟啉配合物06的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体6(34mg,0.046mmol,1.0eq),二氯化铂(37mg,0.14mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=4:1–1:1),得目标产物红色固体15mg,收率35%。1hnmr(500mhz,cdcl3):δ7.77(dd,j=6.5,1.5hz,6h),8.10(dd,j=6.5,1.5hz,2h),8.15(d,j=8.5hz,6h),8.33(dd,j=6.5,2.0hz,2h),8.87(d,j=5.0hz,2h),8.96-8.99(m,6h).其发射光谱图见图2.
实施例7:a3b不对称型铂金属卟啉配合物07的合成
配体7的合成:向干燥100ml三口烧瓶加入磁子,抽换氮气三次。然后加入对甲氧基苯甲醛(304ul,2.5mmol,1.0eq),对氟苯甲醛(805ul,7.5mmol,3.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉319mg,取46mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:3),得目标产物紫色固体21mg,收率8%。1hnmr(500mhz,cdcl3):δ-2.80(s,2h),4.12(s,3h),7.32(dd,j=7.0,2.0hz,2h),7.46-7.49(m,6h),8.14(dd,j=6.5,2.0hz,2h),8.17-8.20(m,6h),8.84(d,j=6.5hz,6h),8.92(d,j=4.5hz,2h).
a3b不对称型铂金属卟啉配合物07的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体7(32mg,0.046mmol,1.0eq),二氯化铂(37mg,0.14mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=4:1–1:1),得目标产物红色固体32mg,收率34%。1hnmr(500mhz,cdcl3):δ4.11(s,3h),7.30(dd,j=7.0,2.0hz,2h),7.44-7.47(m,6h),8.13(dd,j=6.5,2.0hz,2h),8.16-8.19(m,6h),8.93(d,j=6.0hz,6h),9.01(d,j=4.5hz,2h).
实施例8:a3b不对称型铂金属卟啉配合物08的合成
配体8的合成:向干燥100ml三口烧瓶加入磁子,抽换氮气三次。然后加入对甲氧基苯甲醛(304ul,2.5mmol,1.0eq),苯甲醛(765ul,7.5mmol,3.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉111mg,取45mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体19mg,收率3%。1hnmr(500mhz,cdcl3):δ-2.77(s,2h),4.10(s,3h),7.29(dd,j=3.5,2.0hz,2h),7.73-7.80(m,9h),8.13(dd,j=7.0,2.0hz,2h),8.20-8.22(m,6h),8.84(d,j=3.5hz,6h),8.89(d,j=5.0hz,2h).
a3b不对称型铂金属卟啉配合物08的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体8(11mg,0.018mmol,1.0eq),二氯化铂(14mg,0.054mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=3:1-1:1),得目标产物红色固体12.5mg,收率83%。1hnmr(500mhz,cdcl3):δ4.11(s,3h),7.30(dd,j=6.5,2.5hz,2h),7.73-7.80(m,9h),8.14(dd,j=6.5,2.0hz,2h),8.20-8.23(m,6h),8.95(d,j=4.5hz,6h),8.99(d,j=4.5hz,2h).其发射光谱见图3.
实施例9:a3b不对称型铂金属卟啉配合物09的合成
配体9的合成:向带有磁力转子的干燥100ml三口烧瓶中加入对氯苯甲醛(1.05g,7.5mmol,3.0eq)。抽换氮气三次。然后加入对氟苯甲醛(268ul,2.5mmol,1.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉451mg,取48mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体39mg,收率20%。1hnmr(500mhz,cdcl3):δ-2.86(s,2h),7.46(t,j=8.5hz,2h),7.75(d,j=8.5hz,6h),8.13(d,j=8.0hz,6h),8.16(dd,j=8.0,5.5hz,2h),8.84(s,8h).
a3b不对称型铂金属卟啉配合物09的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体9(34mg,0.046mmol,1.0eq),二氯化铂(37mg,0.14mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=4:1-3:1),得目标产物红色固体20mg,收率46%。1hnmr(500mhz,cdcl3):δ7.46-7.50(m,2h),7.77(dt,j=6.5,4.0hz,6h),8.14-8.16(m,6h),8.18-8.21(m,2h),8.86(s,2h),8.96(t,j=1.5hz,6h).其发射光谱见图4.
实施例10:a3b不对称型铂金属卟啉配合物10的合成
配体10的合成:向带有磁力转子的干燥100ml三口烧瓶中加入对氯苯甲醛(1.05g,7.5mmol,3.0eq)。抽换氮气三次。然后加入苯甲醛(255ul,2.5mmol,1.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉377mg,取46mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:1),得目标产物紫色固体45mg,收率20%。1hnmr(500mhz,cdcl3):δ-2.86(t,j=10.5hz,2h),7.73-7.76(m,7h),7.77-7.81(m,2h),8.13-8.15(m,6h),8.19-8.22(m,2h),8.82-8.87(m,8h).
a3b不对称型铂金属卟啉配合物10的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体10(30mg,0.041mmol,1.0eq),二氯化铂(33mg,0.12mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=4:1-2:1),得目标产物红色固体20mg,收率54%。1hnmr(500mhz,cdcl3):δ7.75-7.78(m,7h),7.78-7.81(m,2h),8.14-8.17(m,6h),8.21-8.24(m,2h),8.93-8.99(m,6h).其发射光谱见图5.
实施例11:a3b不对称型铂金属卟啉配合物11的合成
配体11的合成:向干燥100ml三口烧瓶加入磁子,抽换氮气三次。然后加入对甲氧基苯甲醛(304ul,2.5mmol,1.0eq),对甲基苯甲醛(888ul,7.5mmol,3.0eq),三氟乙酸(38ul,0.5mmol,0.2eq),醋酸酐(2.0ml,2.5mmol,8.5eq),吡咯(700ul,10.0mmol,4.0eq),溶剂醋酸和丙酸(各20ml)。然后混合物置于130℃油浴下避光反应1小时,加入硝基苯(2.0ml,20.0mmol,8.0eq),继续在130℃油浴下避光反应1小时。反应体系冷却至常温,加入30ml甲醇溶液,0℃温度下静置12小时,抽滤,采用大量甲醇冲洗,干燥。得粗卟啉257mg,取52mg通过硅胶柱色谱分离纯化,淋洗剂(石油醚/二氯甲烷=3:1-1:1.5),得目标产物紫色固体38mg,收率11%。1hnmr(500mhz,cdcl3):δ-2.77(s,2h),2.71(s,9h),4.10(s,3h),7.29(dd,j=6.5,1.5hz,2h),7.56(d,j=8.0hz,6h),8.10(d,j=7.5hz,6h),8.13(dd,j=6.5,2.0hz,2h),7.86(d,j=3.0hz,8h).
a3b不对称型铂金属卟啉配合物11的合成:向带有磁力转子的干燥100ml三口烧瓶中依次加入卟啉配体11(9mg,0.013mmol,1.0eq),二氯化铂(10mg,0.04mmol,3.0eq)。抽换氮气三次,然后加入苯甲腈(35ml)。然后混合物置于25℃油浴下搅拌反应4小时,升温至190℃,继续反应2小时。冷却至常温,减压旋蒸出苯甲腈,随后通过硅胶柱色谱分离纯化,淋洗剂(石油醚:二氯甲烷=4:1-2:1),得目标产物红色固体11mg,收率97%。1hnmr(500mhz,cdcl3):δ2.71(s,9h),4.11(s,3h),7.29(dd,j=7.0,2.0hz,2h),7.56(d,j=7.5hz,6h),8.10(d,j=8.0hz,6h),8.13(dd,j=6.5,2.0hz,2h),7.86(d,j=4.0hz,8h).其发射光谱见图6
显然,上述实施例仅仅是为清楚地说明本发明的内容所做的举例,而非对实施方式的限定。对于所属领域的技术人员来说,在上述说明的基础之上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以一一举例。而由此所引申的显而易见的变化或变动仍处于本发明的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!