一种氮掺杂碳量子点及其制备方法与应用与流程
本发明涉及功能材料制备技术领域,尤其涉及一种氮掺杂碳量子点及其制备方法与应用。
背景技术:
全氟化合物(perfluorinatedchemicals,简称pfcs)是碳氢化合物及其衍生物中的氢原子全部被氟原子取代后所生成的一类化合物。自1951年由美国3m公司研制成功以来,该化合物就凭借其优良的热稳定性、化学稳定性及疏水疏油等特性,被大量用于化工、纺织、皮革、消防、日用洗涤剂、炊具制造等诸多与人类日常生活息息相关的产业中。环境中存在的两种典型全氟化合物全氟辛磺酸盐(perfluorooctanesulfonate,pfos)和全氟辛酸(perfluorooctaneacid,pfoa)均是多种全氟化合物在环境中转化的最终产物。由于氟是电负性最强的非金属元素,碳-氟共价键有极强的极性和很高的化学键能,使得pfos和pfoa能抵抗各种化学或生化作用,甚至在某些强氧化剂和强酸强碱等极端条件下仍能稳定存在。目前,全世界在地表水、地下水、活性污泥、空气、土壤、海洋水以及所有动植物和人体血清中均检出了pfos,研究者甚至在活体北极熊体内也检测到了高浓度的pfos存在。鉴于其分布广、在生态系统中毒性累积性强、难以在环境中被降解且可以长距离迁移等特性,其安全性受到了国内外研究工作者的一致广泛关注;2009年5月在日内瓦缔约方第四届大会上全氟辛磺酸、全氟辛磺酸盐和全氟辛基磺酰氟被正式列入持久性有机污染物名单加以限制。
虽然环境中pfos和pfoa的含量一般较低,但其可以通过食物链进入活体组织造成生物蓄积和生物放大,它们在人体内的半衰期长达5.4年和3.8年之久,能对生物体全身多部位脏器造成危害,引起人类和动物体产生诸如生殖毒性、神经毒性和肝脏毒性等多种毒性作用。因此,pfcs的广泛应用和潜在健康危害已引起研究者对其在环境中的迁移转化和污染状况的持续关注。近年来,研究人员在开发合适的量化分析方法上做了大量的研究工作。当前,对pfos和pfoa的检测多采用气相色谱-质谱(gc-ms)、毛细管电泳(cze)、液相色谱-质谱(lc-ms)、高效液相色谱-串联质谱(hplc-ms/ms)等仪器分析手段,其中cze的检测灵敏度较低,其它三种质谱法虽具有较高的选择性,但这些方法均需要繁琐的样品前处理如衍生、净化、预富集等步骤,且需要相对较长的分析时间和昂贵的仪器设备,使其大范围应用和推广受限。近来也有采用电化学发光法、光电化学传感器、荧光法等对水中pfos和pfoa进行检测的报道,尤其是其中的荧光法由于灵敏度高、操作简便快速,得到了研究者的青睐,是目前受关注和研究最多的方法。
近年来,我国西南大学的谭克俊研究团队在采用荧光法检测pfos做了一系列创新性研究,但主要仍利用有机荧光显色剂作为标记物。荧光方法虽具有较高的灵敏度,但目前所用有机荧光标记物在实际应用中往往存在毒性大、稳定性差等缺陷而难以应用于进一步的原位分析。因此,提供一种发光强度稳定、对pfos选择性高的材料,具有十分重要的意义。
技术实现要素:
有鉴于此,本发明的目的在于提供一种氮掺杂碳量子点及其制备方法与应用。本发明提供的氮掺杂碳量子点能够与pfos特异性结合,进而引起氮掺杂碳量子点荧光的变化,通过测定氮掺杂碳量子点荧光变化量与pfos浓度之间的关系来测定pfos的含量。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种氮掺杂碳量子点的制备方法,包括以下步骤:
将含有原花青素的粗提物、有机氮掺杂剂和水混合,进行水热碳化反应,得到氮掺杂碳量子点。
优选地,所述有机氮掺杂剂包括三乙胺或乙二胺。
优选地,所述含有原花青素的粗提物中原花青素的质量含量为3~5%。
优选地,所述含有原花青素的粗提物与有机氮掺杂剂的用量比为1~1.25g:2ml。
优选地,制备所述含有原花青素的粗提物的原料包括油茶籽内壳或外壳。
优选地,所述水热碳化反应的温度为180~220℃。
优选地,所述水热碳化反应的时间为6~8h。
本发明还提供了上述技术方案所述制备方法制备得到的氮掺杂碳量子点,所述氮掺杂碳量子点为球形,晶格间距为0.20nm,尺寸分布在3~9nm范围,平均尺寸为5.84nm。
本发明还提供了上述技术方案所述氮掺杂碳量子点在选择性检测水中全氟辛基磺酸盐中的应用。
优选地,所述应用包括以下步骤:将氮掺杂碳量子点、全氟辛磺酸盐和缓冲溶液混合、定容,在370~380nm激发波长下测定体系的荧光光谱。
本发明提供了一种氮掺杂碳量子点的制备方法,包括以下步骤:将含有原花青素的粗提物、有机氮掺杂剂和水混合,进行水热碳化反应,得到氮掺杂碳量子点。本发明以含有原花青素的粗提物为碳前躯体,与有机氮掺杂剂混合再进行水热反应后,形成的氮掺杂碳量子点表面官能团丰富,具有强的荧光和优异的ph稳定性和高离子强度耐受性;对水中全氟辛磺酸盐表现出高选择性。同时,由于原花青素中含有芳香碳和多酚结构,这些基团的存在有利于高发光性能碳点的形成;此外,粗提物中可能还含有少量蛋白质等物质,提供自掺杂氮源,增强发光性能,进一步提高了氮掺杂碳量子点的性能。本发明提供的氮掺杂碳量子点与全氟辛磺酸盐结合迅速,反应可迅速完成,且445nm处的荧光信号常温下保持稳定至少1.5h。从实施例可以看出,本发明的氮掺杂碳量子点对全氟辛磺酸盐的检测线性范围为3~160×10-10mol/l,检出限为0.3nm,可用于环境水样中痕量全氟辛磺酸盐的快速检测。
附图说明
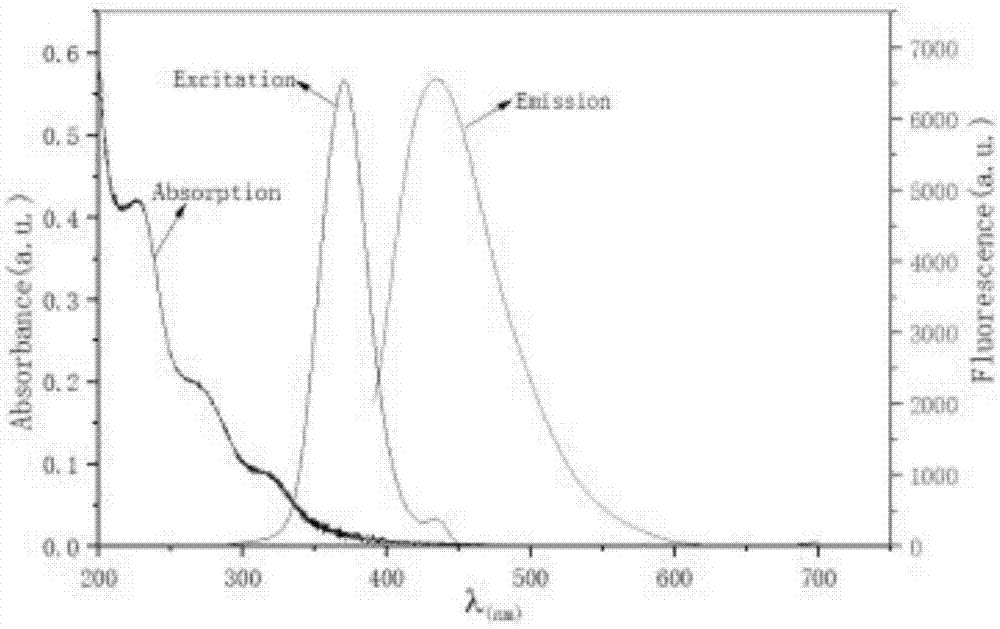
图1为实施例1制备的氮掺杂碳量子点的紫外吸收光谱、最大激发光谱和最大发射光谱;
图2为实施例1制备的氮掺杂碳量子点在不同激发波长下的荧光光谱图;
图3为实施例1制备的氮掺杂碳量子点的傅里叶变换红外光谱图;
图4为实施例1制备的氮掺杂碳量子点的透射电子显微镜照片;
图5为实施例1制备的氮掺杂碳量子点的粒径分布直方图;
图6为实施例1制备的氮掺杂碳量子点在不同ph值下的荧光响应效果图;
图7为实施例1制备的氮掺杂碳量子点在不同浓度nacl溶液中的荧光强度效果图;
图8为不同物质对实施例1制备的氮掺杂碳量子点荧光性质的影响效果图;
图9为不同离子与pfos两者共存的条件下,对实施例1制备的氮掺杂碳量子点荧光的影响效果图;
图10为添加不同浓度pfos时,实施例1制备的氮掺杂碳量子点的荧光光谱图;
图11为对比例1制备的碳量子点的透射电子显微镜照片;
图12为对比例1制备的碳量子点的粒径分布直方图;
图13为对比例1制备的碳量子点的傅里叶变换红外光谱图;
图14为添加不同浓度pfos时,对比例1制备的碳量子点的荧光光谱图。
具体实施方式
本发明提供了一种氮掺杂碳量子点的制备方法,包括以下步骤:
将含有原花青素的粗提物、有机氮掺杂剂和水混合,进行水热碳化反应,得到氮掺杂碳量子点。
在本发明中,所述有机氮掺杂剂优选为三乙胺或乙二胺。在本发明中,所述含有原花青素的粗提物中原花青素的质量含量优选为3~5%,更优选为3.5~4.8%,最优选为4.0~4.5%。在本发明中,所述含有原花青素的粗提物与有机氮掺杂剂的用量比优选为1~1.25g:2ml,更优选为1.05~1.20g:2ml,最优选为1.10~1.15g:2ml。本发明对制备含有原花青素的粗提物的原料没有特殊的限定,采用含有原花青素的物质即可,具体的,如油茶籽内壳或外壳。本发明对所述混合没有特殊的限定,采用本领域技术人员熟知的混合方式即可,具体的,如超声。在本发明中,所述超声的功率优选为50~200w,更优选为80~180w,最优选为100~150w。在本发明中,所述超声的时间优选为10~60min,更优选为20~40min,最优选为30min。
本发明对所述含有原花青素的粗提物的获取方式没有特殊的限定,采用本领域技术人员熟知的含有原花青素的粗提物获取方式即可,具体的,如超声、微波、超声微波联合。在本发明的实施例中,所述含有原花青素的粗提取物的制备方法优选包括以下步骤:(a)称取一定量油茶籽内壳,先用蒸馏水洗净,自然晾干后于40℃真空干燥箱中干燥4h以除去水分,干燥至恒重后取出用粉碎机粉碎并过100目筛,然后分批量将过筛后的油茶壳粉末转入索氏提取器中,加入石油醚,在50℃水浴中加热回流12h脱脂,最后将脱脂后油茶壳粉末自然晾干,密封放于真空干燥箱中,干燥至恒重后,置于干燥瓶中密封保存备用;(b)称取10.0g预处理后的油茶籽内壳,以60%的乙醇水溶液作为提取溶剂,提取溶剂中含有0.1%纤维素酶磷酸盐溶液0.5ml;料液比为1:6(w/v),使用超声-微波协同萃取仪,在超声频率40khz、微波功率300w、提取时间60s和提取温度50℃条件下进行提取;提取液经过滤后,用50℃水浴浓缩蒸干,得到含有原花青素的粗提物。
在本发明中,由于原花青素中含有芳香碳和多酚结构,这些基团的存在有利于高发光性能碳点的形成;此外,粗提物中可能还含有少量蛋白质等物质,提供自掺杂氮源,增强发光性能,进一步提高了氮掺杂碳量子点的性能。
在本发明中,所述水热碳化反应的温度优选为180~220℃,更优选为185~215℃,最优选为190~200℃。在本发明中,升温至水热碳化反应温度的升温速率优选为2~5℃/min,更优选为3~4℃/min。在本发明中,所述水热碳化反应的时间优选为6~8h,更优选为6.5~7.5h,最优选为7h。
水热碳化反应结束后,本发明优选将水热碳化反应产物冷却至室温,经后处理,得到所述氮掺杂碳量子点。在本发明中,所述后处理优选包括以下步骤:冷却后的水热碳化反应产物依次经离心分离和过滤,得到澄清透明的棕色溶液;将澄清透明的棕色溶液浓缩蒸发、冷冻干燥,得到所述氮掺杂碳量子点。在本发明中,所述离心分离的转速优选为9000~11000r/min,更优选为9500~10500r/min,最优选为10000r/min;所述过滤的滤膜优选为0.22μm滤膜;所述浓缩蒸发的温度优选为60℃。在本发明中,所述冷冻干燥的温度优选为-40~-20℃,更优选为-35~-25℃,最优选为-20℃;所述冷冻干燥的时间优选为12h。
本发明还提供了上述技术方案所述制备方法制备得到的氮掺杂碳量子点,所述氮掺杂碳量子点为球形,晶格间距为0.20nm,尺寸分布在3~9nm范围,平均尺寸为5.84nm。
本发明还提供了上述技术方案所述氮掺杂碳量子点在选择性检测水中全氟辛基磺酸盐中的应用。
在本发明中,所述氮掺杂碳量子点应用于选择性检测水中全氟辛基磺酸盐时,优选包括以下步骤:将氮掺杂碳量子点、全氟辛磺酸盐和缓冲溶液混合、定容,在380nm激发波长下测定体系的荧光光谱。在本发明中,所述缓冲溶液优选为br缓冲溶液。在本发明中,所述氮掺杂碳量子点优选以氮掺杂碳量子点溶液的形式使用。在本发明中,所述氮掺杂碳量子点溶液的浓度优选为1~2mg/ml,更优选为1~1.5mg/ml,最优选为1.3mg/ml。在本发明中,所述定容后混合溶液的ph值优选为5~8,更优选为5.5~7.5,最优选为6~7。本发明对所述缓冲溶液的ph值和用量没有特殊的限定,只要能使定容后混合溶液的ph值为5~8即可。
下面结合实施例对本发明提供的氮掺杂碳量子点及其制备方法与应用进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
将1.25g油茶果壳原花青素粗提物、2ml三乙胺和30ml去离子水超声混合,放入50ml反应釜;将反应釜放入鼓风干燥箱,以5℃/min的速度升温至200℃,于200℃进行水热碳化反应6h;反应结束后,将水热碳化反应产物冷却至室温;依次经9000r/min离心分离、过0.22μm水相滤膜过滤,得到澄清透明的棕色溶液;将该溶液于60℃水浴蒸发浓缩至2~3ml,于-40℃冷冻干燥12h,得到氮掺杂碳量子点,命名为n-cds;测定n-cds的荧光量子产率(qy)约为10%。
所述油茶果壳原花青素粗提物的提取过程包括:(a)称取一定量油茶籽内壳,先用蒸馏水洗净,自然晾干后于40℃真空干燥箱中干燥4h以除去水分,干燥至恒重后取出用粉碎机粉碎并过100目筛,然后分批量将过筛后的油茶壳粉末转入索氏提取器中,加入石油醚,在50℃水浴中加热回流12h脱脂,最后将脱脂后油茶壳粉末自然晾干,密封放于真空干燥箱中,干燥至恒重后,置于干燥瓶中密封保存备用;(b)称取10.0g预处理后的油茶籽内壳,以60%的乙醇水溶液作为提取溶剂,提取溶剂中含有0.1%纤维素酶磷酸盐溶液0.5ml;料液比为1:6(w/v),使用超声-微波协同萃取仪,在超声频率40khz、微波功率300w、提取时间60s和提取温度50℃条件下进行提取;提取液经过滤后,用50℃水浴浓缩蒸干,得到含有原花青素的粗提物。
研究n-cds的紫外吸收光谱、最大激发光谱和最大发射光谱,结果如图1所示。从图1可以看出:n-cds在250nm和370nm左右处分别有个典型的紫外吸收峰;在250nm附近的吸收峰由若干小峰组成,归因于苯环的π→π*跃迁和震动效应的重叠;同时,370nm处的吸收峰可归因于n→π*(羧基和/或c-n)转换。n-cds的最大激发和发射波长分别为370nm和450nm。
研究n-cds在不同激发波长下的荧光光谱图,结果如图2所示。从图2可以看出:随着激发波长从300nm变为370nm时,n-cds的发射强度呈现增强趋势,且发射波长稍有蓝移;当激发波长超过370nm,发射波长保持稳定但强度逐渐降低;所制备的n-cds并未像大多数文献报道中那样表现出强烈的激发波长依赖性,表明所制备碳点的粒径分布比较窄,与粒径分布结果一致。
在nexus670红外傅里叶变换光谱仪上使用kbr压片法研究n-cds的傅里叶变换红外光谱图,结果如图3所示。从图3可以看出,所制备n-cds表面主要含有-oh,-nh+,-nh2+,c-nh-c,c=o,c=s,coo-及芳环等基团,说明n-cds表面含有丰富的官能团,提高了n-cds的结合力。
使用场发射透射电子显微镜来研究n-cds的形态和结构,结果如图4所示。从图4可以看出:n-cds具有球形形状,并且很好地分散在水溶液中;此外,碳点的晶格条纹清晰可见(图4插图),晶格间距为0.20nm,对应于石墨(sp2)碳的(102)衍射面。
根据n-cds的场发射透射电子显微镜图片,总结出n-cds的粒径分布直方图,结果如图5所示,从图5可以看出,n-cds的粒径分布比较窄,尺寸在3~9nm范围内分布,平均尺寸5.84nm。
称取1.0086gn-cds溶于30ml去离子水中作为n-cds储备液,以供后续测试使用。
研究n-cds在不同ph值下的荧光稳定性:取一系列0.4ml的n-cds储备液置于一系列10ml比色管中,再分别加入1mlph为3.2~10.5的br缓冲溶液,用去离子水定容至刻度,在370nm激发下,测定450nm下的荧光强度,结果见图6。从图6可以看出:在370nm激发时450nm处n-cds的荧光强度在宽范围的ph值(3.21~10.46)下强且稳定。
研究n-cds在不同浓度nacl溶液中的荧光稳定性:取一系列0.4ml的n-cds储备液溶液置于一系列10ml比色管中,再分别加入一系列不同浓度的nacl溶液,用去离子水定容至刻度,在370nm激发下,测定450nm下的荧光强度,结果见图7。从图7可以看出:在不同浓度的nacl存在下,n-cds强度基本稳定,在高浓度(1m)nacl的存在下荧光强度约降低10%,表明高离子强度环境下,n-cds具有高稳定性。
为了探索n-cds作为荧光探针的选择性,研究了环境中常见离子、pfos和几种结构相似物对n-cds荧光性质的影响,包括agno3,bacl2,nacl,al(no3)3,cdso4,mnso4,feso4,coso4,k2cr2o7,cr(no3)3,k2cro4,ca(no3)2,pfoa,pfos,全氟丁基磺酸盐,全氟己烷磺酸盐;其中重金属盐的设定浓度为1×10-6m,其余物质浓度为1×10-4m;实验结果如图8所示,在本实验中所检测的所有物质中,n-cds对pfos有良好的选择性。
同时,还探究了在不同离子共存下对pfos与n-cds结合体系的荧光强度影响,实验结果如图9所示。从图9可以看出:共存物的存在对pfos与n-cds结合体系的荧光响应强度基本无明显影响。综上所述,n-cds对pfos具有良好的选择性。
将本实施例制备的氮掺杂碳量子点应用于检测水中pfos,具体步骤为:在添加1mlph值为10.8的br缓冲溶液条件下,加入一系列浓度的pfos溶液到0.4mln-cds储备液中,用去离子水定容到10ml,在380nm激发波长下测定体系的荧光光谱,结果如图10所示,图10中,荧光光谱图从底部到顶部分别代表在n-cds储备液中添加0、3、9、10、30、50、70、90、100、140、160×10-10mpfos时n-cds的荧光光谱。从图10可以看出:添加pfos后,443nm处荧光强度随着pfos浓度的增大而增大;荧光增加量(δf)和pfos浓度在3~160×10-10mol/l范围内呈线性关系(r2=0.9987)(见插入图),其中δf为pfos存在和不存在条件下的体系荧光的差值;方法检测限为0.3nm。
从当地采集自来水、江水和湖水,所采集的水样用0.22μm有机滤膜(聚偏氟乙烯滤膜)过滤并储存在预先清洗干燥后的锥形瓶中;同样,取0.4mln-cds储备液于10ml比色管中,然后加入1mlbr缓冲溶液,用采集的水样定容至刻度线,在380nm激发波长下测定实际水样体系的荧光强度。通过添加不同浓度的pfos标准样品研究其加标回收率,用于评价所构建荧光探针用于环境水样中目标物检测的可行性。通过加标回收实验分析,将所构建的n-cds探针应用于实际水样中的pfos的检测,结果列于表1中。从表1可以看出,三种水样中添加已知量的pfos的回收率分别在91.60~105.28%,94.9~109.9%和95.2~101.02%;相对标准偏差(rsd)分别在1.36~5.8%,1.13~5.64%和1.09~4.6%范围之内;表明所构建n-cds可作为实际环境水样分析中直接测定pfos的优良荧光探针。
表1不同环境水样中pfos的加标回收测定结果
实施例2
将1.25g油茶果壳原花青素粗提物、2ml三乙胺和30ml去离子水超声混合,放入50ml反应釜;将反应釜放入鼓风干燥箱,以2℃/min的速度升温至180℃,于180℃进行水热碳化反应8h;反应结束后,将水热碳化反应产物冷却至室温;依次经9000r/min离心分离、过0.22μm水相滤膜过滤,得到澄清透明的棕色溶液;将澄清透明的棕色溶液于60℃水浴蒸发浓缩至2~3ml,于-40℃冷冻干燥12h,得到n-cds;测定n-cds的荧光量子产率(qy)为7%。
所述油茶果壳原花青素粗提物的提取过程与实施例1相同。
实施例3
将1.25g油茶果壳原花青素粗提物、2ml乙二胺和30ml去离子水超声混合,放入50ml反应釜;将反应釜放入鼓风干燥箱,以5℃/min的速度升温至200℃,于200℃进行水热碳化反应7h;反应结束后,将水热碳化反应产物冷却至室温;依次经9000r/min离心分离、过0.22μm水相滤膜过滤,得到澄清透明的棕色溶液;将澄清透明的棕色溶液于60℃水浴蒸发浓缩至2~3ml,于-40℃冷冻干燥12h,得到n-cds。测定n-cds的荧光量子产率(qy)约为5%。
所述油茶果壳原花青素粗提物的提取过程与实施例1相同。
对比例1
将1.25g油茶果壳原花青素粗提物和30ml去离子水超声混合,放入50ml反应釜;将反应釜放入鼓风干燥箱,以5℃/min的速度升温至180℃,于180℃进行水热碳化反应6h;反应结束后,将水热碳化反应产物冷却至室温;依次经9000r/min离心分离、过0.22μm水相滤膜过滤,得到澄清透明的棕色溶液;将澄清透明的棕色溶液于60℃水浴蒸发浓缩至2~3ml,冷冻干燥12h,得到碳量子点;测定碳量子点的荧光量子产率(qy)为6%。
所述油茶果壳原花青素粗提物的提取过程与实施例1相同。
使用场发射透射电子显微镜来研究碳量子点的形态和结构,结果如图11从图11可以看出,碳量子点稳定性差。
根据碳量子点的场发射透射电子显微镜图片,总结出碳量子点的粒径分布直方图,结果如图12所示,从图12可以看出,碳量子点尺寸较大且粒径分布范围宽。
在nexus670红外傅里叶变换光谱仪上使用kbr压制丸粒研究碳量子点的傅里叶变换红外光谱图,结果如图13所示。从图13可以看出:未见氨基离子及c-s等相关峰。
将对比例1制备的碳量子点应用于检测水中pfos,具体步骤为:称取1.0086g碳量子点溶于30ml去离子水中作为碳量子点储备液;在添加缓冲溶液条件下,加入一系列浓度的pfos溶液到0.4ml碳量子点储备液中,用去离子水定容到10ml,在380nm激发波长下测定体系的荧光光谱,结果如图14所示。图14中,荧光光谱图从底部到顶部分别代表在碳量子点储备液中添加0.04、0.1、0.4、0.6、0.8、1.0和1.2×10-6m时碳量子点的荧光光谱。从图14可以看出:测定线性范围窄为0.04~1.2×10-6mol/l,灵敏度低,检出限为3.16×10-8mol/l。
从实施例可以看出,本发明的氮掺杂碳量子点对全氟辛磺酸盐的检测线性范围为3~160×10-10mol/l,检出限为0.3nm,可用于环境水样中痕量全氟辛磺酸盐的快速检测。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!